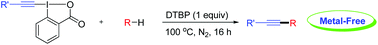Tout sur les médicaments הכל על תרופות كل شيئ عن الأدوية Все о наркотиках 关于药品的一切 డ్రగ్స్ గురించి అన్ని 마약에 관한 모든 것 Όλα για τα Ναρκωτικά Complete Tracking of Drugs Across the World by Dr Anthony Melvin Crasto, Worldpeacepeaker, worlddrugtracker, PH.D (ICT), MUMBAI, INDIA, Worlddrugtracker, Helping millions, 9 million hits on google on all websites, 2.5 lakh connections on all networks, “ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
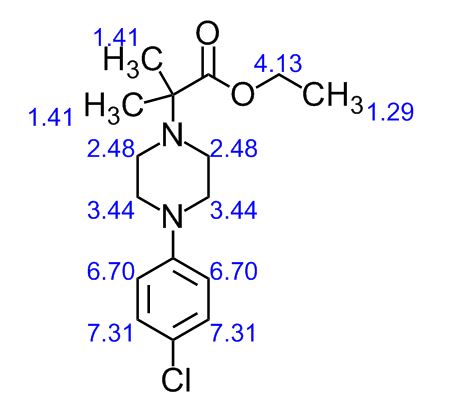
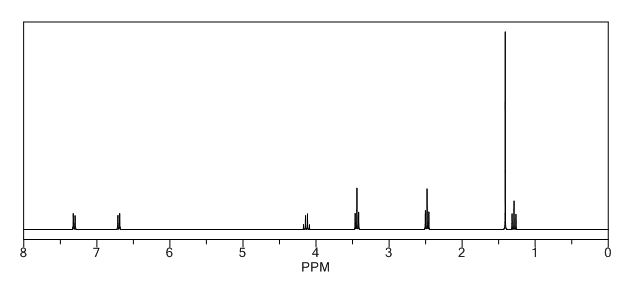
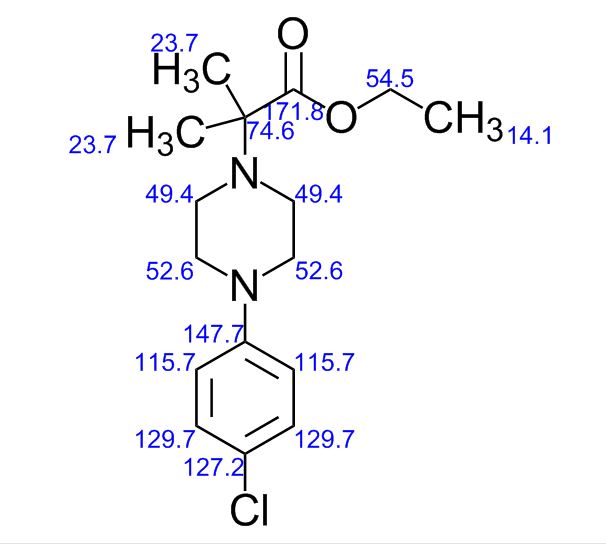
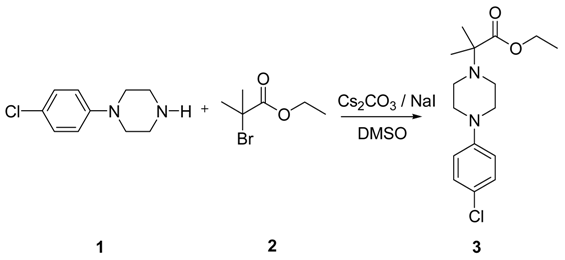
To a solution of 4-(4-chlorophenyl)piperazine dihydrochloride 1 (5.0 g, 0.0185 mol) in DMSO (30 ml), anhydrous cesium carbonate (30.0 g, 0.0925 mol), sodium iodide (1.39 g, 0.0093 mol) and ethyl 2-bromo-2-methylpropanoate 2 (3.97 g, 0.02 mol) were added. The resulting mixture was stirred at 25-30oC for 12 hours. The reaction mass was diluted with water (200 ml) and extracted with ethyl acetate (2 x 200 ml). The ethyl acetate layer was washed with water (2 x 100 ml), dried over anhydrous sodium sulfate (10.0 g) and concentrated under vacuum. The crude product thus obtained was purified by column chromatography (stationary phase silica gel 60-120 mesh; mobile phase 10% ethyl acetate in hexane). The title compound 3 was obtained as a white solid (4.73 g, 82 %).
1Department of Chemistry, Sambalpur University, JyotiVihar-768019, Orissa, India
2Institute of Chemical Technology (ICT), Matunga, Mumbai-400019, Maharashtra, India
*Author to whom correspondence should be addressed.
Received: 17 May 2009 / Accepted: 30 June 2009 / Published: 27 July 2009
Bijay K Mishra
Professor at Sambalpur University, Chemistry Department
Abstract
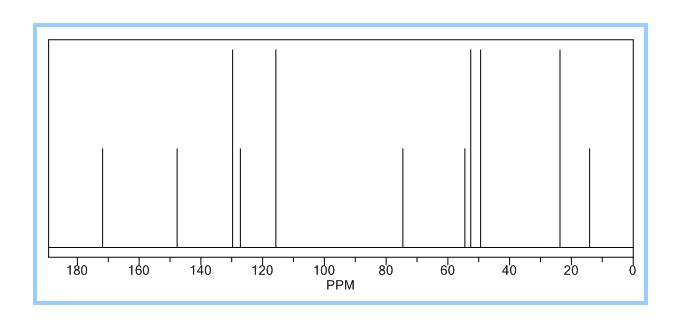
The title compound was synthesized by N-alkylation of 4-(4-chlorophenyl)piperazine with ethyl 2-bromo-2-methylpropanoate and its IR, 1H NMR, 13C NMR and Mass spectroscopic data are reported.
Preparation of 1-(2-Methoxyphenoxy)-2,3-epoxypropane 4.
To a stirring solution of 2-methoxy phenol 2 (10 kg, 80.55 mol) and water (40 L) at about 30 °C was added sodium hydroxide (1.61 kg, 40.25 mol) and water (10 L). After stirring for 30−45 min, epichlorohydrin 3 (22.35 kg, 241.62 mol) was added and stirred for 10−12 h at 25−35 °C. Layers were separated, and water (40 L) was added to the organic layer (bottom layer) containing product. Sodium hydroxide solution (3.22 kg, 80.5 mol) and water (10 L) were added at 27 °C and stirred for 5−6 h at 27 °C.
The bottom product layer was separated and washed with sodium hydroxide solution (3.0 kg 75 mol) and water (30 L). Excess epichlorohydrin (3) was recovered by distillation of the product layer at below 90 °C under vacuum (650−700 mmHg) to give 13.65 kg (94%) of title compound with 98.3% purity by HPLC, 0.2% of 2- methoxy phenol 2, 0.1% of epichlorohydrin 3, 0.1% of chlorohydrin 11, 0.3% of dimer 12 and 0.3% of dihydroxy 13.
PROCESS, SYNTHESIS, UncategorizedComments Off on Development and Manufacturing GMP Scale-Up of a Continuous Ir-Catalyzed Homogeneous Reductive Amination Reaction
The design, development, and scale up of a continuous iridium-catalyzed homogeneous high pressure reductive amination reaction to produce 6, the penultimate intermediate in Lilly’s CETP inhibitor evacetrapib, is described. The scope of this report involves initial batch chemistry screening at milligram scale through the development process leading to full-scale production in manufacturing under GMP conditions. Key aspects in this process include a description of drivers for developing a continuous process over existing well-defined batch approaches, manufacturing setup, and approaches toward key quality and regulatory questions such as batch definition, the use of process analytics, start up and shutdown waste, “in control” versus “at steady state”, lot genealogy and deviation boundaries, fluctuations, and diverting. The fully developed continuous reaction operated for 24 days during a primary stability campaign and produced over 2 MT of the penultimate intermediate in 95% yield after batch workup, crystallization, and isolation.
Development and Manufacturing GMP Scale-Up of a Continuous Ir-Catalyzed Homogeneous Reductive Amination Reaction
ACS Editors’ Choice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes.
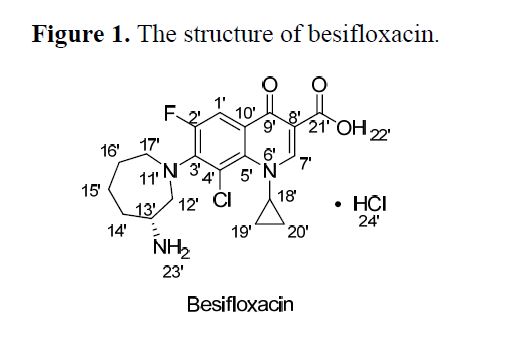
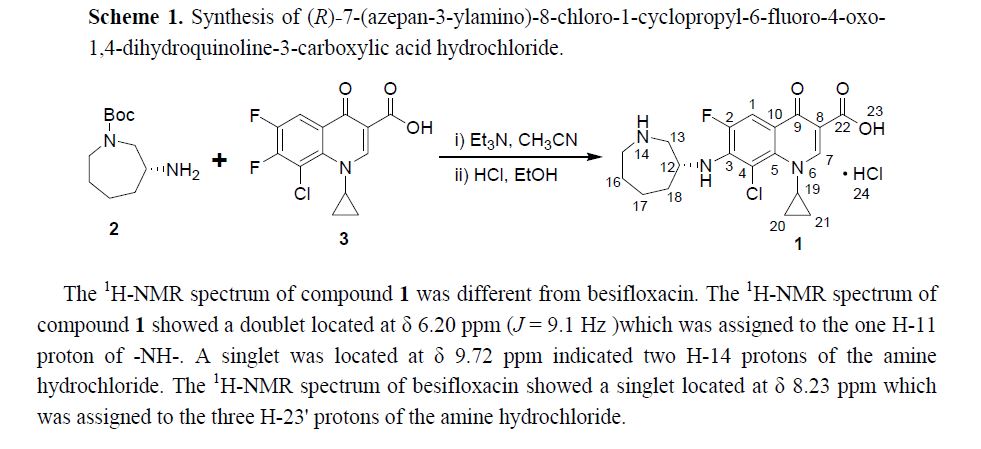
Abstract: In this paper (R)-7-(azepan-3-ylamino)-8-chloro-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid hydrochloride 1 was isolated and identified as the N-substituted regioisomer of besifloxacin, which has been synthesized from the reaction of 8-chloro-1-cyclopropyl-6,7-difluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid 3 with (R)-tert-butyl 3-aminoazepane-1-carboxylate 2 in acetonitrile as solvent in 37% yield. The chemical structure of compound 1 was established on the basis of 1H-NMR, 13C-NMR, mass spectrometry data and elemental analysis.
Structural Characterization
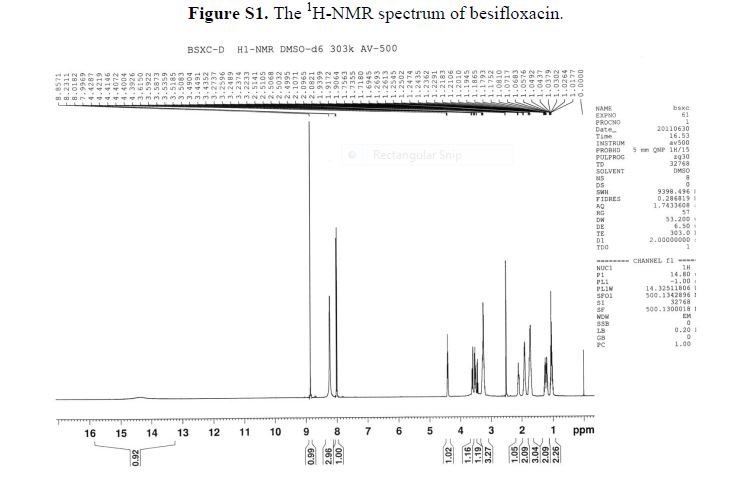
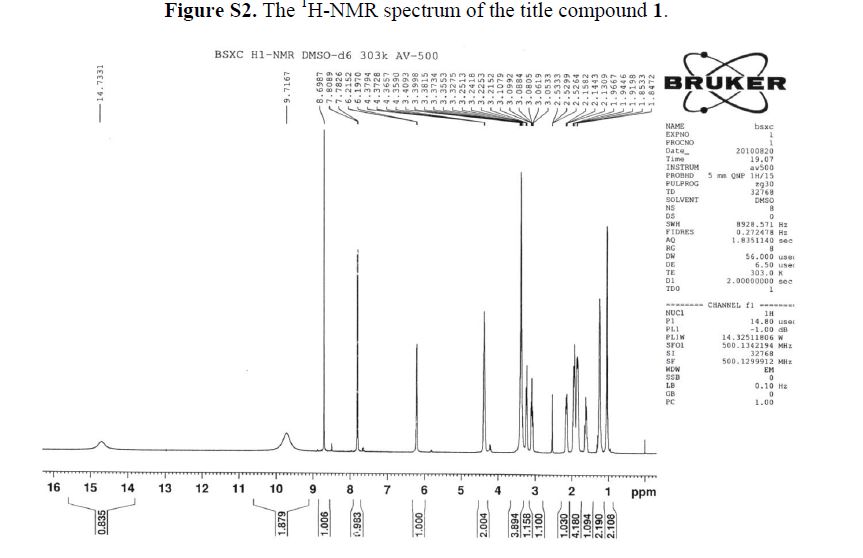
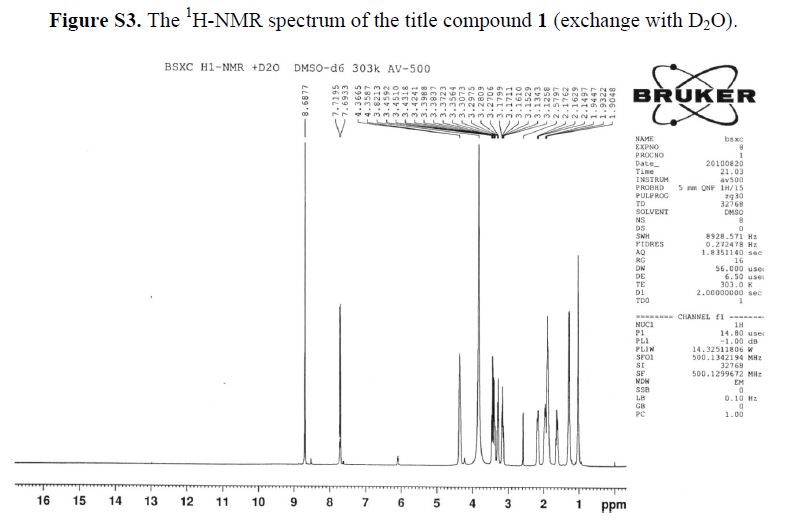
1H-NMR (500 MHz, DMSO-d6): δ ppm: 14.73 (H-23, s, 1H), 9.72 (H-14, s, 2H), 8.69 (H-7, s, 1H),7.79 (H-1, d, J = 13.1 Hz, 1H), 6.20 (H-11, d, J = 9.1 Hz, 1H), 4.37 (H-12 and H-19, m, 2H), 3.38(H-13, m, 2H), 3.23 (H-15, m, 1H), 3.09 (H-15, m, 1H), 2.14 (H-18, m, 1H), 1.94 (H-16 and H-18, m,2H), 1.84 (H-16 and H-17, m, 2H), 1.60 (H-17, m, 1H), 1.23 (H-20 or H-21, m, 2H), 1.03 (H-20 orH-21, m, 2H).
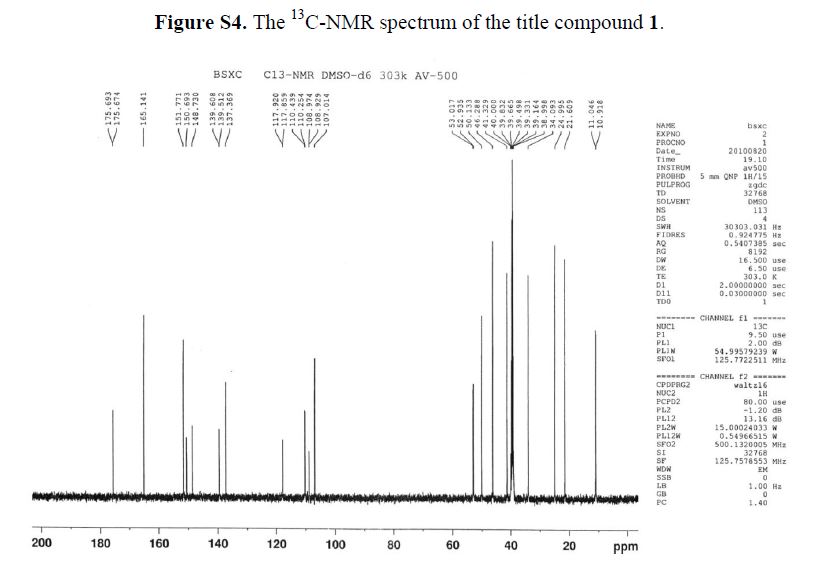
13C-NMR(125 MHz, DMSO-d6): δ ppm: 175.6 (C-9), 165.4 (C-22), 151.7 (C-7), 150.6 (C-2), 148.7(C-3), 139.0 (C-5), 137.3 (C-4), 117.8 (C-10), 110.3 (C-1), 107.0 (C-8), 52.9 (C-12), 50.1 (C-13), 46.2(C-15), 41.3 (C-19), 34.0 (C-18), 24.9 (C-16), 21.6 (C-17), 10.9 (C-20 or C-21).
FAB-MS, m/z = 394.1 (M+).
Elemental analysis: Calculated for C19H21ClFN3O3.HCl: C, 53.03%; H, 5.15%; N, 9.77%; found: C,52.82%; H, 5.39%; N, 9.50%.
1H-NMR (500 MHz, DMSO-d6): δ ppm: 14.73 (H-23, s, 1H), 9.72 (H-14, s, 2H), 8.69 (H-7, s, 1H), 7.79 (H-1, d, J = 13.1 Hz, 1H), 6.20 (H-11, d, J = 9.1 Hz, 1H), 4.37 (H-12 and H-19, m, 2H), 3.38 (H-13, m, 2H), 3.23 (H-15, m, 1H), 3.09 (H-15, m, 1H), 2.14 (H-18, m, 1H), 1.94 (H-16 and H-18, m,2H), 1.84 (H-16 and H-17, m, 2H), 1.60 (H-17, m, 1H), 1.23 (H-20 or H-21, m, 2H), 1.03 (H-20 orH-21, m, 2H).
R&D Center, Jiangsu Yabang Pharmaceutical Group, Changzhou 213200, China
In this paper (R)-7-(azepan-3-ylamino)-8-chloro-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid hydrochloride 1was isolated and identified as the N-substituted regioisomer of besifloxacin, which has been synthesized from the reaction of 8-chloro-1-cyclopropyl-6,7-difluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid 3 with (R)-tert-butyl 3-aminoazepane-1-carboxylate 2in acetonitrile as solvent in 37% yield. The chemical structure of compound 1 was established on the basis of 1H-NMR, 13C-NMR, mass spectrometry data and elemental analysis
REGIOMER OF BESIFLOXACIN
BESIFLOXACIN
Zaixin Chen *
R&D Center, Jiangsu Yabang Pharmaceutical Group, Changzhou 213200, China
* Author to whom correspondence should be addressed;
E-Mail: zaixin_chen@163.com.
Zai-Xin Chen
Director of R&D Center at Jiangsu Yabang Pharmaceutical Group Co., Ltd
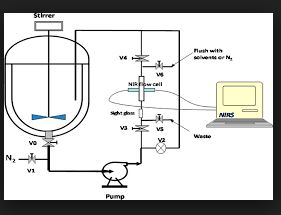
The transamination-chemistry-based process for sitagliptin is a through-process, which challenges the crystallization of the active pharmaceutical ingredient (API) in a batch stream composed of multiple components. Risk-assessment-based design of experiment (DoE) studies of particle size distribution (PSD) and crystallization showed that the final API PSD strongly depends on the seeding-point temperature, which in turn relies on the solution composition.
To determine the solution composition, near-infrared (NIR) methods had been developed with partial least squares (PLS) regression on spectra of simulated process samples whose compositions were made by spiking each pure component, either sitagliptin free base (FB), water, isopropyl alcohol (IPA), dimethyl sulfoxide (DMSO), or isopropyl acetate (IPAc), into the process stream according to a DoE. An additional update to the PLS models was made by incorporating the matrix difference between simulated samples in lab and factory batches.
Overall, at temperatures of 20–35 °C, the NIR models provided a standard error of prediction (SEP) of less than 0.23 wt % for FB in 10.56–32.91 wt %, 0.22 wt % for DMSO in 3.77–19.18 wt %, 0.32 wt % for IPAc in 0.00–5.70 wt %, and 0.23 wt % for water in 11.20–28.58 wt %. After passing the performance qualification, these on-line NIR methods were successfully established and applied for the on-line analysis of production batches for compositions prior to the seeding point of sitagliptin crystallization.
Application of On-Line NIR for Process Control during the Manufacture of Sitagliptin
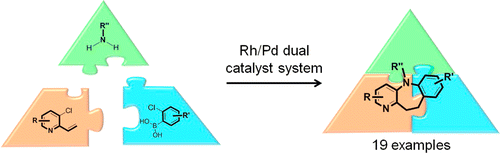
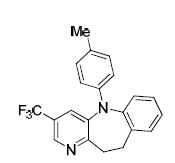
A RhI/Pd0 catalyst system was applied to the multicomponent synthesis of aza-dibenzazepines from vinylpyridines, arylboronic acids, and amines in a domino process with no intermediate isolation or purification.
An efficient and versatile synthesis of chiral tetralins has been developed using both inter- and intramolecular Friedel-Crafts alkylation as a key step. The readily available hydronaphthalene substrates were prepared via a highly enantioselective metal-catalyzed ring opening of meso-oxabicyclic alkenes followed by hydrogenation. A wide variety of complex tetracyclic compounds have been isolated…more
One-Pot Synthesis of Chiral Dihydrobenzofuran Framework via Rh/Pd Catlaysis
Organic Letters
October 12, 2012
A one-pot synthesis of the chiral dihydrobenzofuran framework is described. The method utilizes Rh-catalyzed asymmetric ring opening (ARO) and Pd-catalyzed C-O coupling to furnish the product in excellent enantioselectivity without isolation of intermediates. Systematic metal-ligand studies were carried out to investigate the compatibility of each catalytic system using product enantiopurity as an…more
A game of dominoes: A synthetic route to aza-dihydrodibenzoxepines is described, through the combination of a Rh-catalyzed arylation and a Pd-catalyzed C-O coupling in a single pot. For the first time, the ability to incorporate a chiral and an achiral ligand in a two-component, two-metal transformation is achieved, giving the products in moderate to good yields, with excellent enantioselectivities.
A Rh(I)/Pd(0) catalyst system was applied to the multicomponent synthesis of aza-dibenzazepines from vinylpyridines, arylboronic acids, and amines in a domino process with no intermediate isolation or purification.
Formation of substituted oxa- and azarhodacyclobutanes.
Chemistry – A European Journal
December 6, 2013
The preparation of substituted oxa- and azarhodacyclobutanes is reported. After exchange of ethylene with a variety of unsymmetrically and symmetrically substituted alkenes, the corresponding rhodium-olefin complexes were oxidized with H2O2 and PhINTs (Ts=p-toluenesulfonyl) to yield the substituted oxa- and azarhodacyclobutanes, respectively. Oxarhodacyclobutanes could be prepared with excellent…more
Alexander von Humboldt Foundation
Berlin, Aachen and Gottingen
2009-2014
Visiting Professor
University of Berlin
2009
Visiting Professor
Université de Marseilles
2008
ICIQ Summer School
ICIQ Tarragona, Spain
2008
Attilio Corbella Summer School Professor
Italian Chemical Society
2007
Arthur C. Cope Scholar Award
American Chemical Society
2006
Alfred Bader Award
Canadian Society for Chemistry
2006
R. U. Lemieux Award
Canadian Society for Chemistry
2004
Solvias Prize
Solvias AG
2002
Fellow of the Royal Society of Canada
Royal Society of Canada
2001
Areas of Research Interest and Expertise
new synthetic methods
metal catalyzed cycloaddition and annulation reactions
asymmetric catalysis with focus on rhodium, nickel and palladium catalysts
cyclopropane synthesis and reactions
hydrometallation reactions
reactions of organosilicon and organotin compounds
fragmentation reactions
new routes to medicinally/biologically interesting compounds
heterocycle synthesis using metal catalysts
///////Multicomponent, Multicatalyst Reactions, (MC)2R, Dibenzazepine Synthesis, Mark Lautens, University of Toronto ,
Toronto, Ontario, Jennifer Tsoung
spectroscopy, SYNTHESISComments Off on Continuous Processing and Efficient in Situ Reaction Monitoring of a Hypervalent Iodine(III) Mediated Cyclopropanation Using Benchtop NMR Spectroscopy
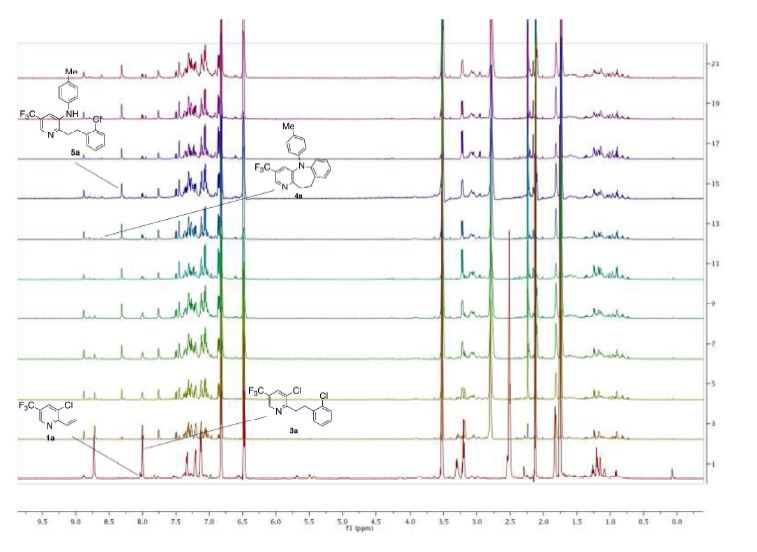
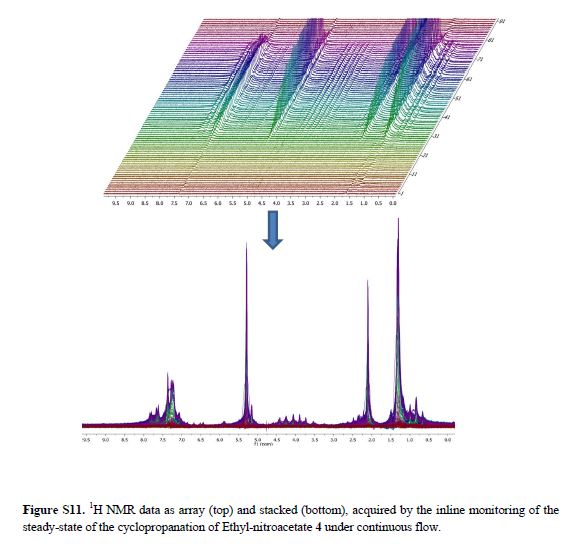
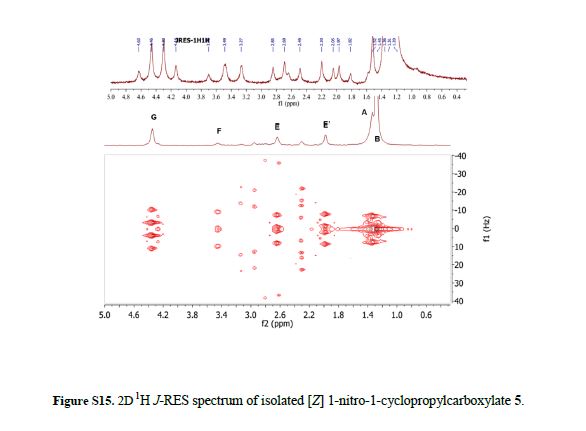
Real-time NMR spectroscopy has proven to be a rapid and an effective monitoring tool to study the hypervalent iodine(III) mediated cyclopropanation. With the ever increasing number of new synthetic methods for carbon–carbon bond formation, the NMR in situ monitoring of reactions is becoming a highly desirable enabling method. In this study, we have demonstrated the versatility of benchtop NMR using inline and online real-time monitoring methods to access mutually complementary information for process understanding, and we developed new approaches for real-time monitoring addressing challenges associated with better integration into continuous processes.
Continuous Processing and Efficient in Situ Reaction Monitoring of a Hypervalent Iodine(III) Mediated Cyclopropanation Using Benchtop NMR Spectroscopy
Steven V. Ley received his PhD from Loughborough University in 1972, after which he carried out post-doctoral research with Professor Leo Paquette at Ohio State University, followed by Professor Derek Barton at Imperial College London. In 1975, he joined that Department as a lecturer and became Head of Department in 1989. In 1992, he moved to the 1702 BP Chair of Organic Chemistry at the University of Cambridge and became a Fellow of Trinity College. He was elected to the Royal Society in 1990 and was President of the Royal Society of Chemistry (RSC) 2000-02. Steve has been the recipient of many prizes and awards including the Yamada-Koga Prize, Nagoya Gold Medal, ACS Award for Creative Work in Synthetic Organic Chemistry and the Paul Karrer Medal.
Flow Chemistry Society – India Chapter is assisting the proliferation of Process Intensification and Flow Chemistry across the country
After an enthusiastic response at the 2nd FCS-India Symposium & Workshop held at IICT-Hyderabad in June’16 with 27companies and 115 delegates attending, we are happy to announce :
The 3rd2-day FLOW CHEMISTRY Symposium + DEMO Workshop is organized on 27th – 28th August 2016 at IISER – PUNE by Flow Chemistry Society – India Chapter (in collaboration with IISER-Pune, NCL & IIT-B) , with speakers & demonstrators from India, UK, Netherlands and Hungary.
Prof. Ashwini Kumar Nangia, Director – CSIR-NCL has kindly consented to be the Hon. Chief Guest and inaugurate the Symposium & Workshop.
Both days have intensive interactive sessions on the theory and industrial applications of Flow Chemistry followed by live–demonstrationsusing 5 to 6 different Flow Reactor platforms –each day from microliters to 10,000 L/day industrial scale.
The Fees are Rs. 7,000 for Industry Delegates and Rs. 3,000 for Academic Delegates
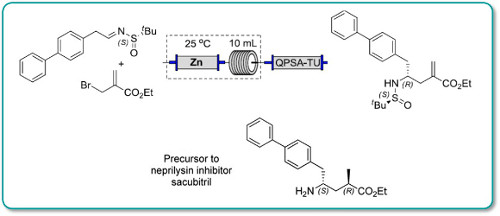
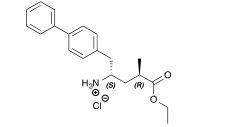
An efficient preparation of a precursor to the neprilysin inhibitor sacubitril is described. The convergent synthesis features a diastereoselective Reformatsky-type carbethoxyallylation and a rhodium-catalyzed stereoselective hydrogenation for installation of the two key stereocenters. Moreover, by integrating machine-assisted methods with batch processes, this procedure allows a safe and rapid production of the key intermediates which are promptly transformed to the target molecule (3·HCl) over 7 steps in 54% overall yield.
Synthesis of a Precursor to Sacubitril Using Enabling Technologies
Continuous flow methodologyhas been used to enhance several steps in the synthesis of a precursor to Sacubitril.
In particular, a key carboethoxyallylation benefited from a reducedprocessing time and improved reproducibility, the latter attributable toavoiding the use of a slurry as in the batch procedure. Moreover, in batchexothermic formation of the organozinc species resulted in the formation ofside products, whereas this could be avoided in flow because heat dissipationfrom a narrow packed column of zinc was more efficient
Synthesis of a Precursor to Sacubitril Using Enabling Technologies
LCZ696 (sacubitril/valsartan) is a first-in-class combination of the angiotensin II receptor-blocker valsartan and the neprilysin inhibitor sacubitril. A recent head-to-head comparison of LCZ696 with enalapril in a double-blind trial was stopped early because the boundary for an overwhelming benefit with LCZ696 was crossed.As a result of this, LCZ696 was reviewed under the FDA’s priority review program and was granted approval on the July 7, 2015 to reduce the risk of cardiovascular death and hospitalization for HF in patients with chronic HF (NYHA Class II–IV) and reduced ejection fraction.
LCZ696 is a complex aggregate comprised of the anionic forms of sacubitril and valsartan, sodium cations, and water molecules in the molar ratio of 1:1:3:2.5, respectively
To a stirred solution of (2R, 4S)-5-(4-Biphenylyl)-2-methyl-4-(tert-butylsulfinylamino)valeric acid 14 (50.0 mg, 134 μmol) in absolute ethanol (0.4 mL) at 0 °C was added thionyl chloride (20 μL, 268 μmol). The reaction mixture was stirred at room temperature for 3 h. The solvent was removed to yield 46.0 mg (99%) of titled compound 3 as a white solid.
Zhi-Fei Cheng, Yi-Si Feng, Chun Rong, Tao Xu, Peng-Fei Wang, Jun Xu, Jian-Jun Dai, Hua-Jian Xu
A general and efficient alkynylation of unactivated C(sp3)-H bonds under metal-free conditions was developed herein.
Directed alkynylation of unactivated C(sp3)–H bonds with ethynylbenziodoxolones mediated by DTBP
aSchool of Chemistry and Chemical Engineering, School of Biological and Medical Engineering, Hefei University of Technology, Hefei 230009, P. R. China
bAnhui Key Laboratory of Controllable Chemical Reaction and Material Chemical Engineering, Hefei 230009, P. R. China E-mail: hjxu@hfut.edu.cn Fax: (+86)-551-62904405
cAnhui Provincial Laboratory of Heterocyclic Chemistry, Maanshan 243110, China
A general and efficient method for the direct alkynylation of unactivated C(sp3)–H bonds under metal-free conditions is described. The reaction performs smoothly under mild conditions and shows excellent functional-group tolerance. Initial mechanistic investigation indicates that the reaction may involve a radical pathway.
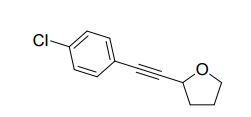
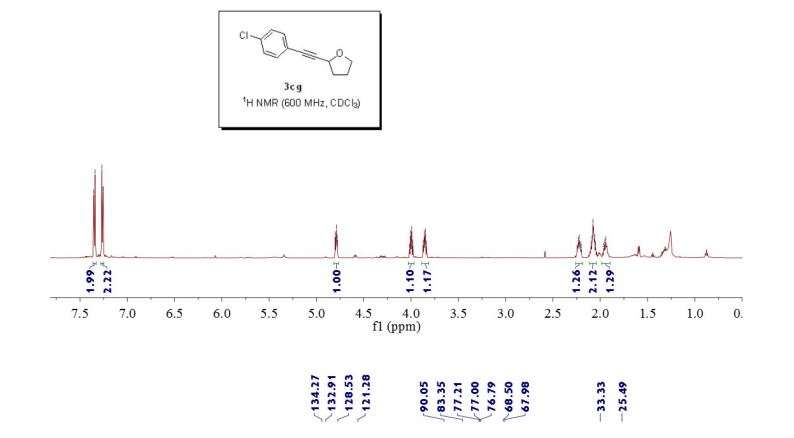
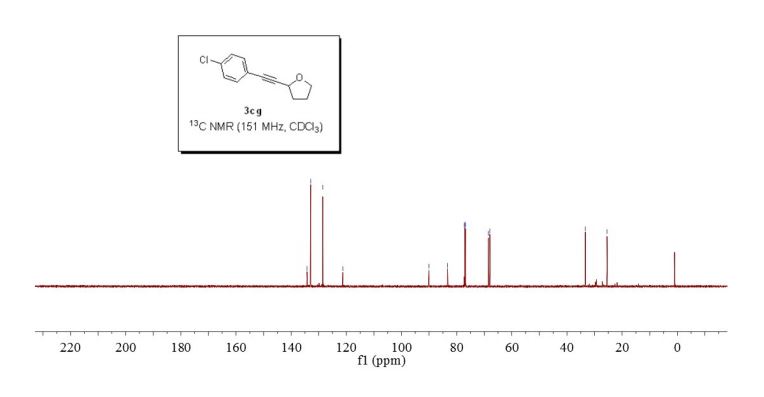
2-((4-chlorophenyl)ethynyl)tetrahydrofuran (3cg) ref 1 : Following general procedure, The product was purified by flash column chromatography on silica gel (petroleum ether) and 1c : 2g = 1:69, obtained in 70 % yield as a pale yellow oil (28.8 mg).



























 BESIFLOXACIN
BESIFLOXACIN