












the isolated cyclopentenedione derivative may have structure 1a or 1b or even exist as an equilibrium mixture between these two enol forms showing average 1H and 13C NMR spectra due to a proposed rapid interconversion between 1a and 1b.
http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0103-50532005000300024

Synthetic results
Our approach to cyclopentenedione derivative (1) started with the preparation of furylmethylcarbinol (3) by the reduction of commercially available 2-acetylfuran (2) with NaBH4 (Scheme 2).5 Compound 3 was isolated in 98% yield and transformed into 4-hydroxy-5-methylcyclopenten-2-one (4) in 90% yield after treatment with ZnCl2-HCl (pH 6.0) under reflux in dioxane-H2O for 48 h.6 Upon treatment of 4-hydroxy-5-methylcyclopenten-2-one (4) with phosphate buffer (pH 8.0) in refluxing dioxane for 24 h, 4-hydroxy-2-methylcyclopenten-2-one (5) was obtained in 65% yield.7 By using this strategy we were able to prepare up to gram quantities of hydroxyketone 5.

Diketone 6 was obtained in almost quantitative yield by the smooth oxidation of hydroxyketone 5 with MnO2(Scheme 3).8,9 At this point, all that remained was to carry out the necessary acylation coupling. It was with some gratification that we observed that the reaction between lithium enolate of diketone 6 and cinnamic anhydride 7 gave a 57:43 mixture of cyclopentenediones 1a/1b in 22% yield, after purification by flash column chromatography, together with starting material and by-products arising from O-acylation (Scheme 3).



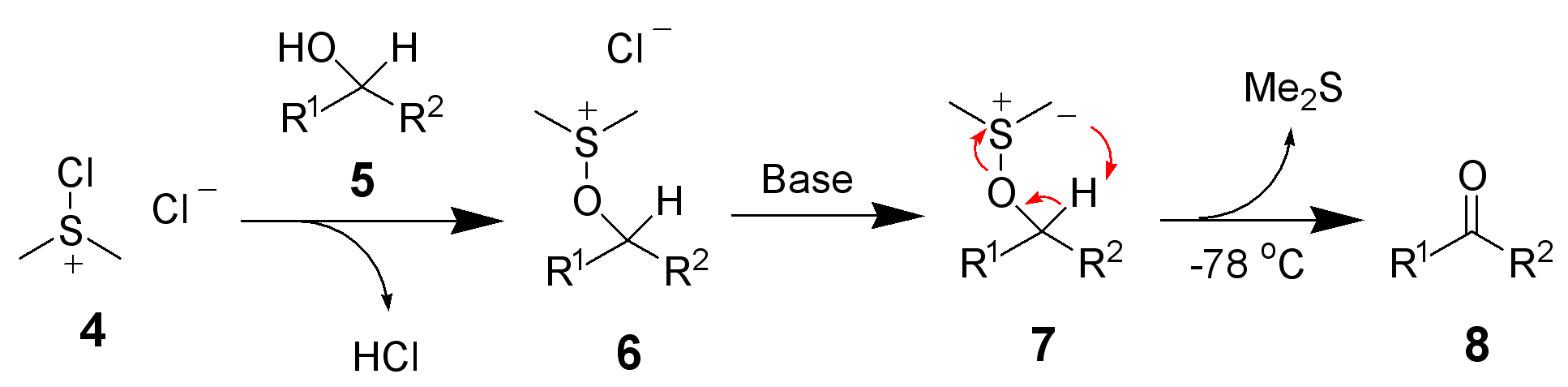
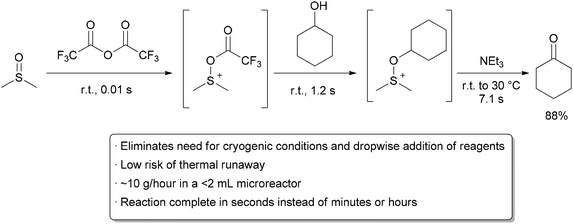
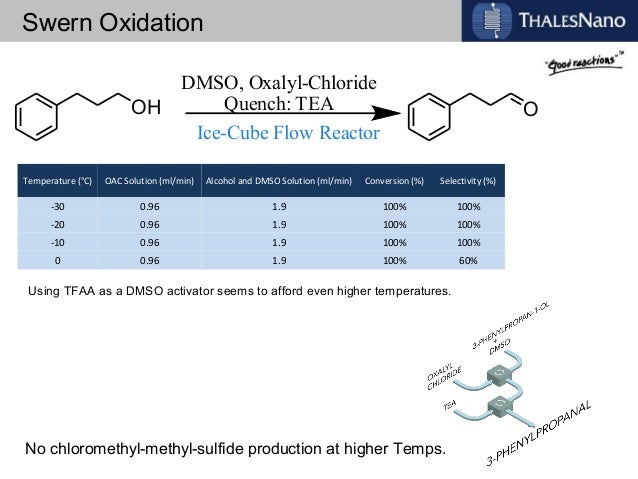
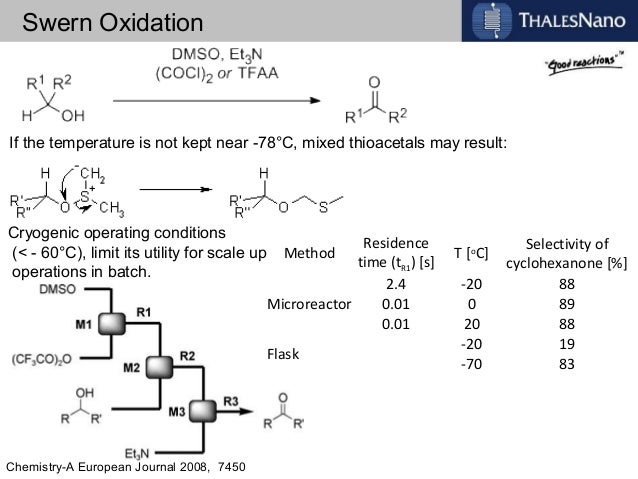
In order to try to improve the yields for formation of 1a/1b, we tested a new synthetic route (Scheme 4). Protection of the OH-functionality in 5 with TESCl and imidazole at room temperature gave ketone 8 in 85% yield. Treatment of 8 with LDA in THF at –78 ºC, followed by slow addition of cinnamaldehyde, gave aldol adduct 9 as a mixture of diastereoisomers. Oxidation of the OH-function at C9 in allylic alcohol 9 under standard Swern11 conditions followed by removal of the TES protecting group with TBAF in THF led to diol 10 in 60% overall yield. The last step involved treatment of diol 10 under standard Swern oxidation conditions, to give a 59:41 mixture of 1a/1b in 79% yield.11
The correct structure for the natural product was confirmed as being 1a by the heteronuclear long-range coupling (nJCH; n = 2,3,4) obtained by HMBC experiments in CDCl3 as solvent. Heteronuclear long-range coupling of C11 (dC 201.3) with H13 (dH 6.70, 3JCH) and H15 (dH 2.12, 3JCH), as well as between C14 (dC 191.8) with H13 (dH 6.70, 2JCH) and H15 (dH 2.12, 4JCH) for 1a, together with the long-range coupling of C11 (dC 200.7) with H12 (dH 6.61, 2JCH) and H15 (dH 2.11, 4JCH), as well as between C14 (dC 192.3) with H12 (dH 6.61, 3JCH) and H15 (dH 2.11 ppm, 3JCH) for 1b, unambiguously established the correct structure as being 1a (Figure 10).

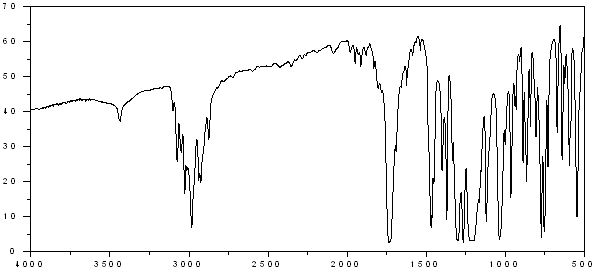
cyclopentenedione derivative (1) as a yellow solid. Rf 0.37 (30% EtOAc/Hexane); IR (film) nmax/cm-1: 3428, 2965, 1632, 1589, 1266, 1103, 1023, 803, 742, 699; (HRMS) Exact mass calc. for C15H12O3: 240.0786. Found: 240.0787.
Journal of the Brazilian Chemical Society
On-line version ISSN 1678-4790
J. Braz. Chem. Soc. vol.16 no.3a São Paulo May/June 2005
http://dx.doi.org/10.1590/S0103-50532005000300024
Short synthesis of a new cyclopentene-1,3-dione derivative isolated from Piper carniconnectivum
Luiz C. Dias*; Simone B. Shimokomaki; Robson T. Shiota
http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0103-50532005000300024
Instituto de Química, Universidade Estadual de Campinas, CP 6154, 13083-970 Campinas – SP, Brazil
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SON
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।










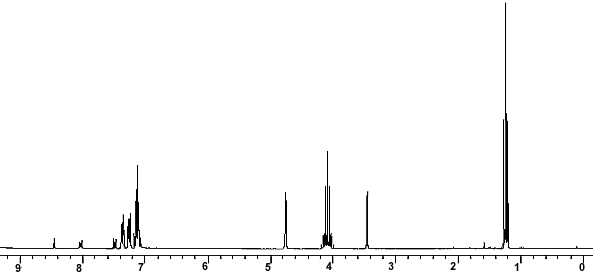
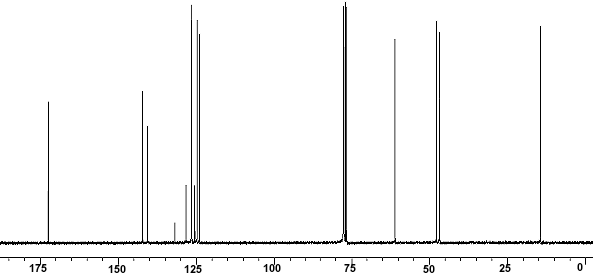
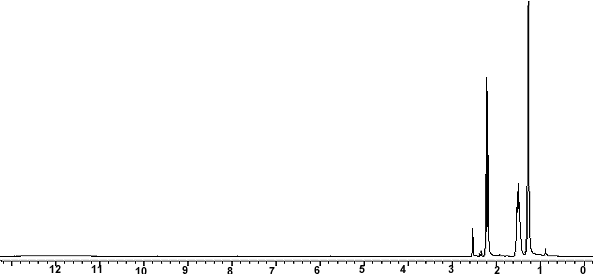
 ppm, CDCl3) 0.30 (m, 1H), 0.54-0.95 (m, 10H), 1.05-1.2 (m, 1H ), 1.39 (s, 9H), 1.89 (d, 3H J 6.8Hz), 2.86 (bs, 1H), 3.28-3.60 (m 2H ), 5.79 (s,1H), 6.24 (d,1H J 15Hz), 6.87 (s 1H), 7.0-7.15 (m 1H), 7.56-7.76 (m 4H), 7.88 (d 1H J 7.85 Hz), 7.92-8.04 (m 2H), 8.68 (d 1H J 8.25 Hz), 8.73 (d 1H J 8.25Hz); 13C NMR (
ppm, CDCl3) 0.30 (m, 1H), 0.54-0.95 (m, 10H), 1.05-1.2 (m, 1H ), 1.39 (s, 9H), 1.89 (d, 3H J 6.8Hz), 2.86 (bs, 1H), 3.28-3.60 (m 2H ), 5.79 (s,1H), 6.24 (d,1H J 15Hz), 6.87 (s 1H), 7.0-7.15 (m 1H), 7.56-7.76 (m 4H), 7.88 (d 1H J 7.85 Hz), 7.92-8.04 (m 2H), 8.68 (d 1H J 8.25 Hz), 8.73 (d 1H J 8.25Hz); 13C NMR (
















 .
.



























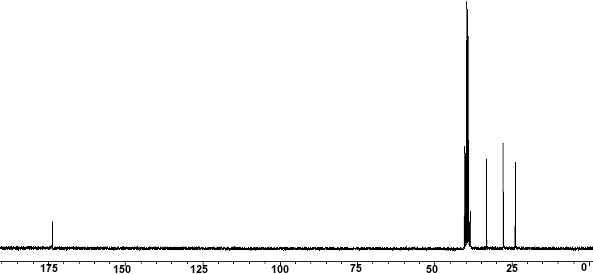
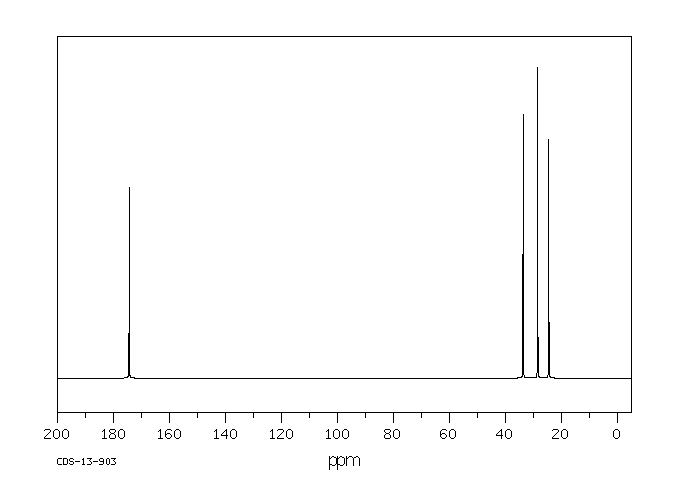
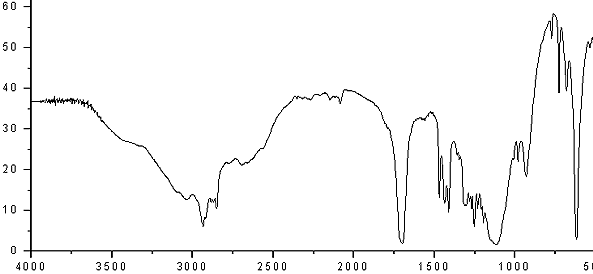
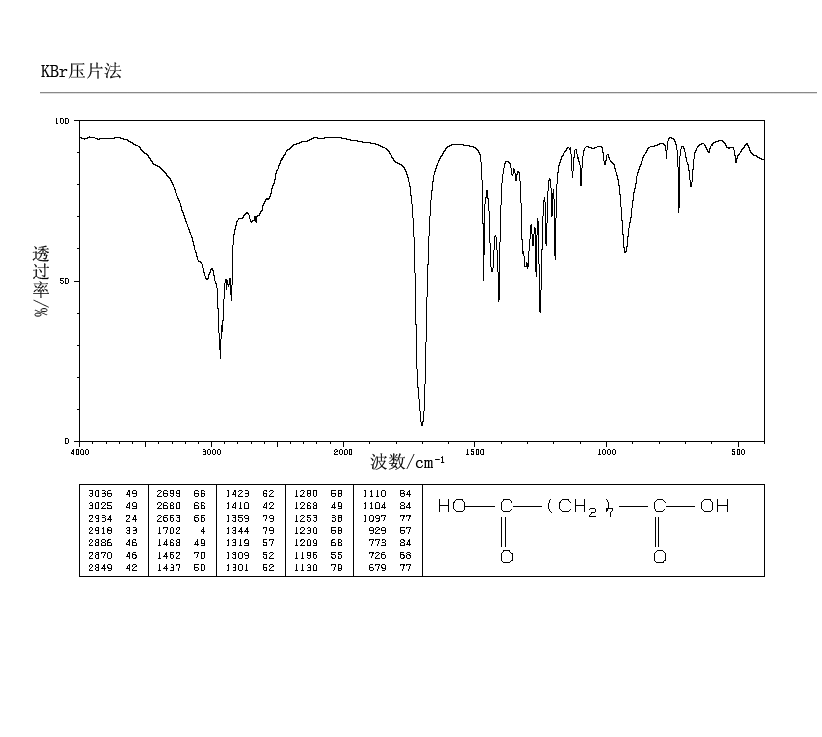










 .
.









 yu rao
yu rao




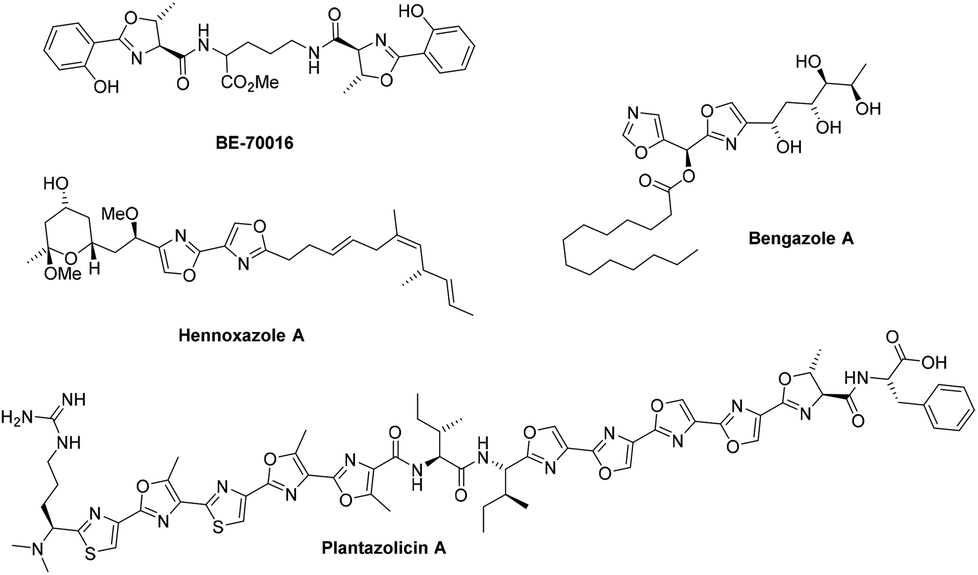
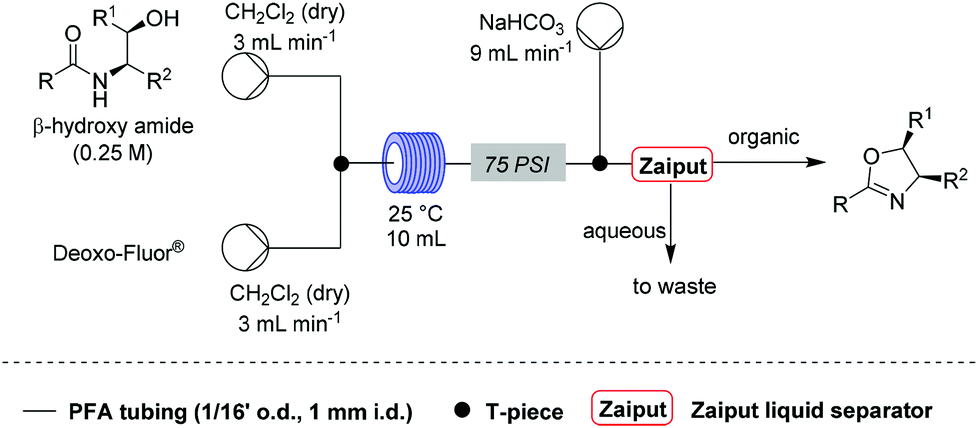
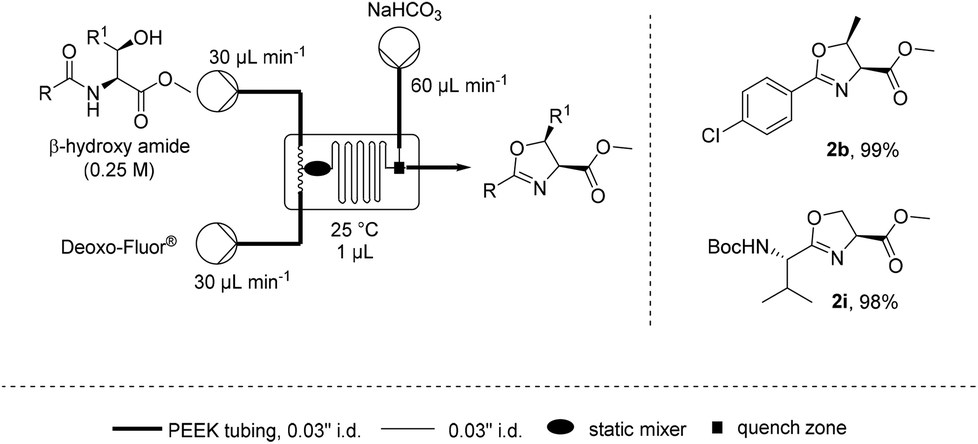
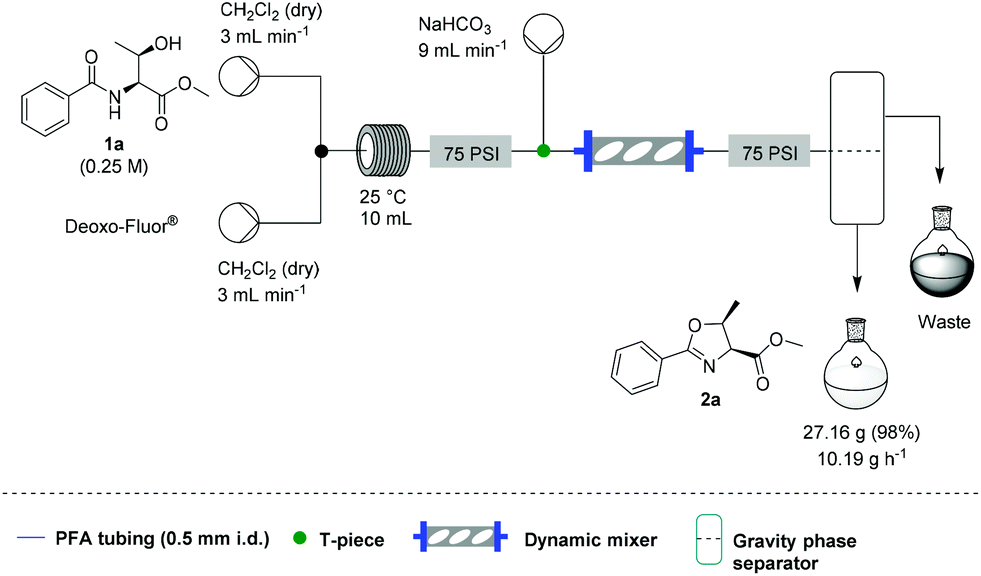
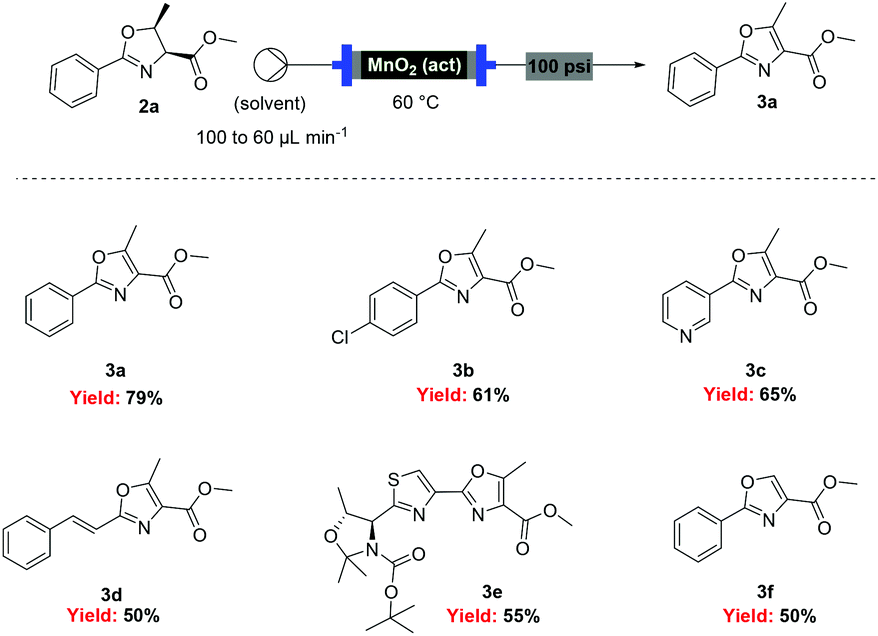
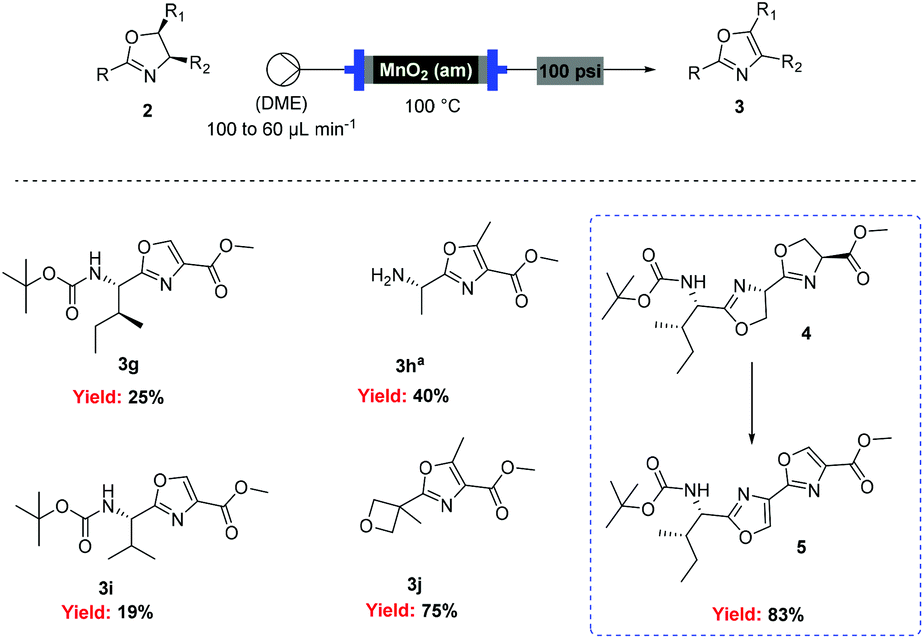
![[thin space (1/6-em)]](http://www.rsc.org/images/entities/char_2009.gif) Deprotection was observed.
Deprotection was observed.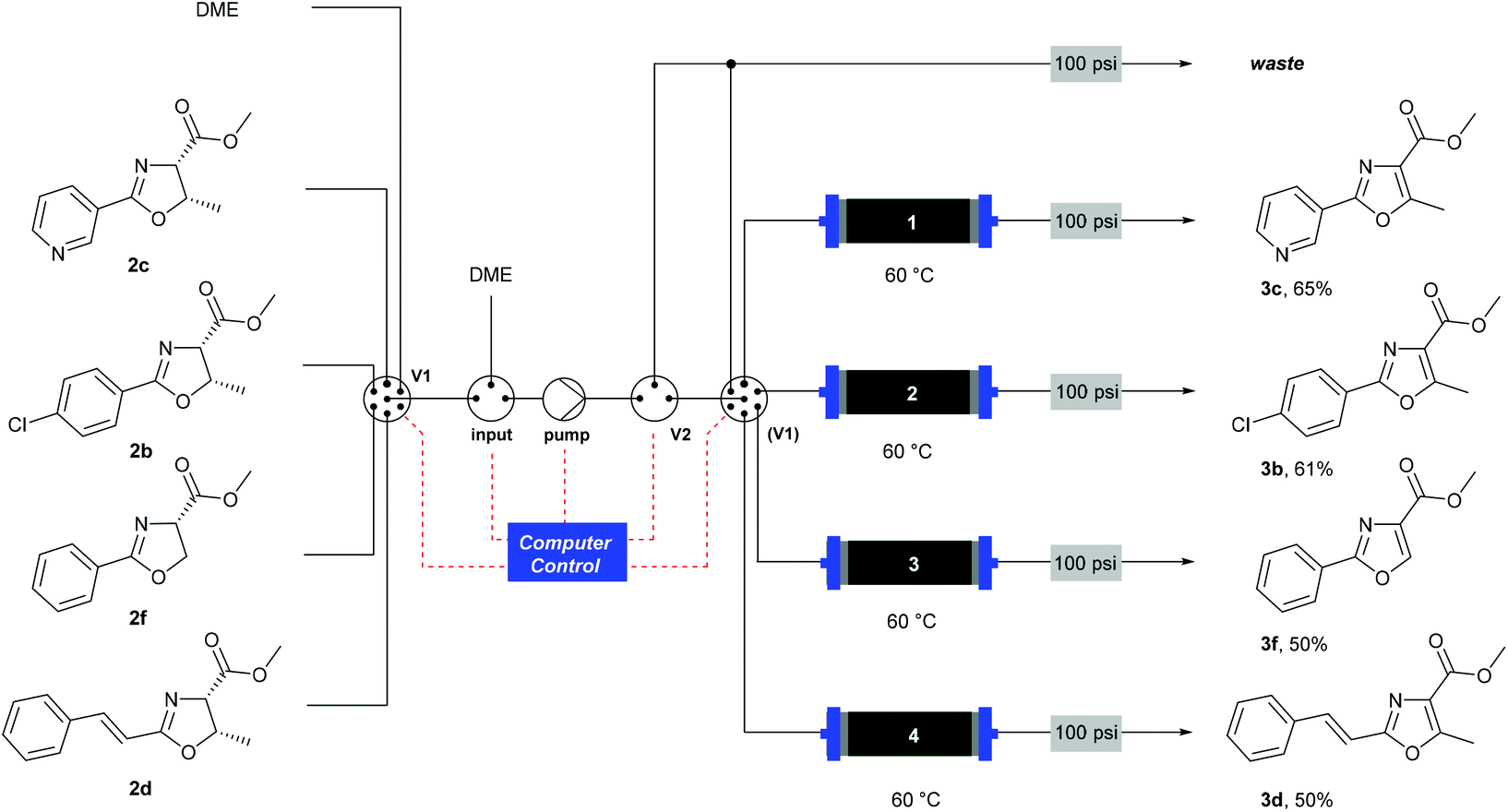
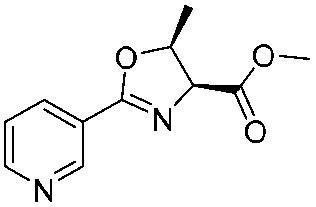
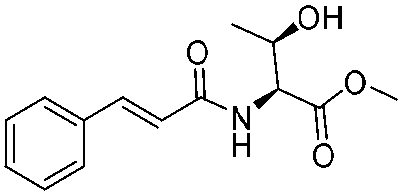
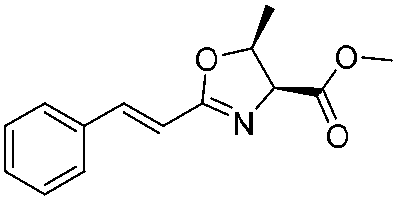
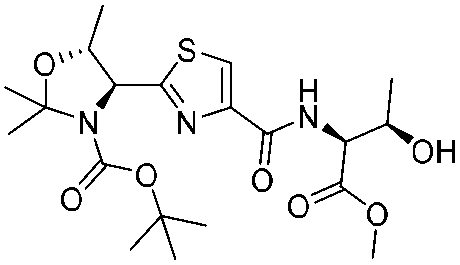
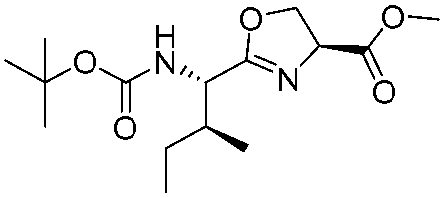
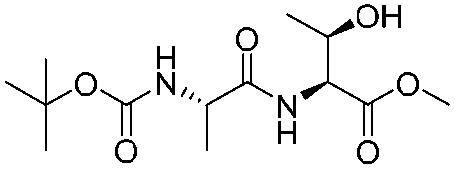
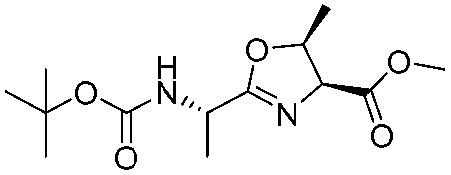
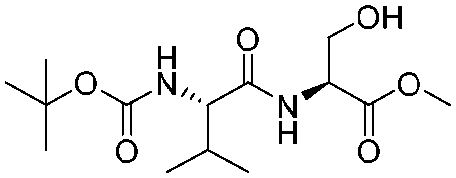
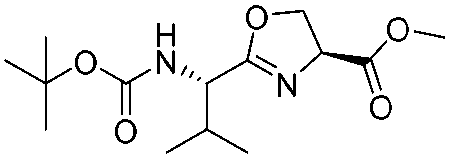
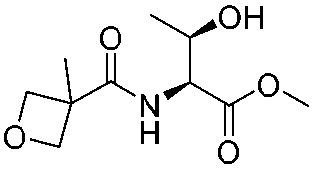
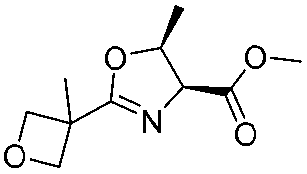
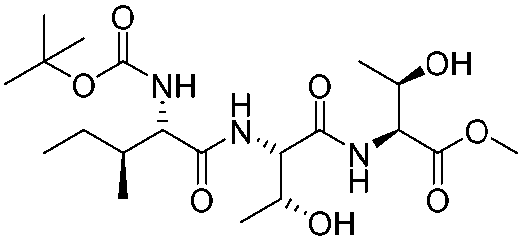
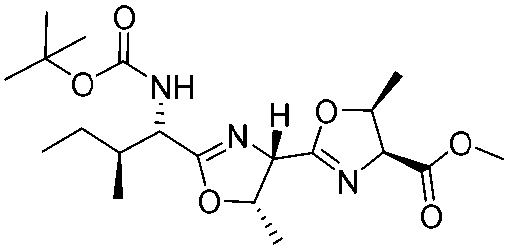


![[small nu, Greek, tilde]](http://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1) = 2953, 1736, 1645, 1603, 1580, 1496, 1450, 1384, 1349, 1244, 1197, 1174, 1067, 1045, 1001, 973, 934, 904, 886, 851, 778, 695;
(cm−1) = 2953, 1736, 1645, 1603, 1580, 1496, 1450, 1384, 1349, 1244, 1197, 1174, 1067, 1045, 1001, 973, 934, 904, 886, 851, 778, 695; 



 .Prof. Dr. Beate Koksch
.Prof. Dr. Beate Koksch fu-berlin.de
fu-berlin.de .
.


") An Anopheles female mosquito that transmits malaria(© picture alliance/dpa Fotografia)Over one million people die of malaria each year because they do not have access to effective drugs.Millions, especially in the developing world, cannot afford the combination drug preparation, which consists mainly of artemisinin.
An Anopheles female mosquito that transmits malaria(© picture alliance/dpa Fotografia)Over one million people die of malaria each year because they do not have access to effective drugs.Millions, especially in the developing world, cannot afford the combination drug preparation, which consists mainly of artemisinin.") Dr. Peter H. Seeberger, Director at the Max Planck Institute of Colloids and Interfaces in Potsdam and Professor of Chemistry at the Freie Universität Berlin(© dpa)“Photochemistry is a simple and cost-effective method. However, the pharmaceutical industry has not used it to date because it was so difficult to control and implement on a large scale,” explains Peter Seeberger.
Dr. Peter H. Seeberger, Director at the Max Planck Institute of Colloids and Interfaces in Potsdam and Professor of Chemistry at the Freie Universität Berlin(© dpa)“Photochemistry is a simple and cost-effective method. However, the pharmaceutical industry has not used it to date because it was so difficult to control and implement on a large scale,” explains Peter Seeberger.



 h
h .
.











{kind=link}
{kind=link}
{kind=link}