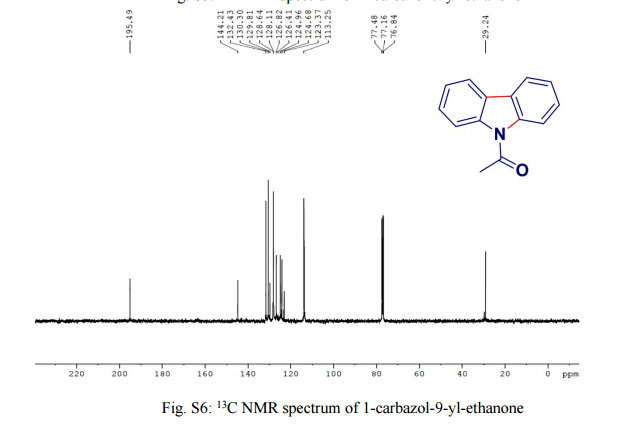
Safe, small scale access to supercritical fluids
The ability to safely access high temperatures and pressures in flow reactors has implications not only on the rate of chemical reactions, but also on the types of solvents one can use. Many greensolvents such as methanol and acetone have boiling points too low for certain batch applications, whereas performing reactions at high pressure in a flow reactor may allow for their safe use at elevated temperatures.
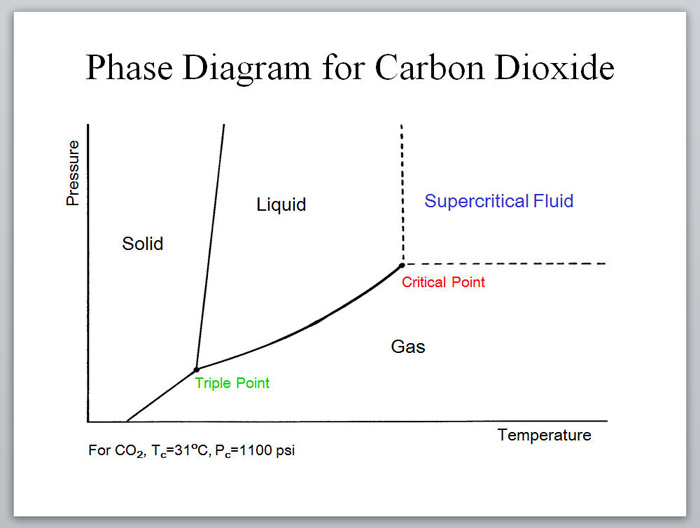
Supercritical fluids are particularly interesting, since these solvents are entirely inaccessible without high pressure conditions. The use of supercritical fluids in a flow system offers numerous advantages over batch reactors.
Reactions may be performed on a small scale, improving safety and reducing the amount of material required. Depending on the type of reactor, it may be possible to visualize the reaction to evaluate the phase behaviour. Moreover, the reaction can be analyzed and the temperature and pressure subsequently changed without stopping the reaction and cleaning the vessel, as is necessary in a simple autoclave.
Continuous methods for utilizing supercritical fluids for extraction,1 chromatography,2 and as a reaction medium3 have all been commercialized, particularly for supercritical carbon dioxide (scCO2).4 Academic examples using scMeOH, scH2O, and scCO2 for continuous reactions such as hydrogenations, esterifications, oxidations, and Friedel–Crafts reactions have been reported.5
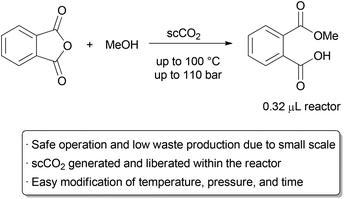
A recent example that illustrates many of the green advantages of performing supercritical fluid chemistry in flow is in the ring opening of phthalic anhydride with methanol by Verboom and co-workers (Scheme 1).6 They designed a microreactor with a volume of just 0.32 μL that can withstand very high pressures.
The exceptionally small channel causes a large build-up of pressure, and supercritical conditions with pressures of up to 110 bar and temperatures up to 100 °C can occur inside the reactor, giving an ‘on-chip’ phase transition. The channel size increases near the outlet, allowing the fluid to expand to atmospheric conditions.
Thus, the total volume of scCO2 under high pressure is exceptionally small, alleviating the major hazards of operating under supercritical conditions. The reaction was thoroughly studied on this small scale, allowing the authors to determine rate constants at several different temperatures and pressures.

 |
|
Scheme 1 Small scale continuous use of supercritical fluids. |
|
Near- and supercritical water (scH2O) can be an interesting green solvent only obtainable at very high temperature (Tc = 374 °C) and pressure (Pc = 221 bar). It is commonly used for completeoxidation of organic waste materials to CO2; however, it has also been shown to be an effective solvent for selective oxidations.7 Given the harshness of the reaction conditions, it is not surprising that side product formation is common and highly dependent on the reaction time. For fast reactions in a batch reactor, precise control of reaction time is challenging, as the vessel takes time to heat and cool. In contrast, rapid heating, cooling, and quenching can be accomplished in a continuous process, allowing for well defined reaction times.
Fine tuning of the temperature, pressure, and time is also easier in a continuous process, as these variables can be changed without stopping and starting the reaction between samples. Thus, more data points can be obtained with less material and fewer heating and cooling cycles.
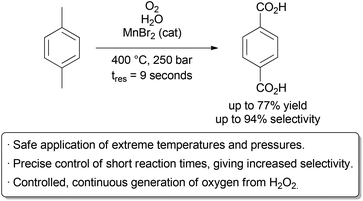
The Poliakoff group used these advantageous to perform a detailed study on the oxidation of p-xylene to terephthalic acid in scH2O, a reaction carried out on industrial scale in acetic acid (Scheme 2).8 By using a flow reactor, reaction times as low as 9 seconds could be used. The equivalents of oxygen could also be finely varied on a small scale through the controlled thermal decomposition of H2O2.
Studying this aerobic oxidation with such precision in a batch process would prove highly challenging. Under optimal conditions, excellent selectivity for the desired product could be obtained. Further research by the same group identified improved conditions for this transformation.9

 |
|
Scheme 2 Selective oxidation in supercritical water. |

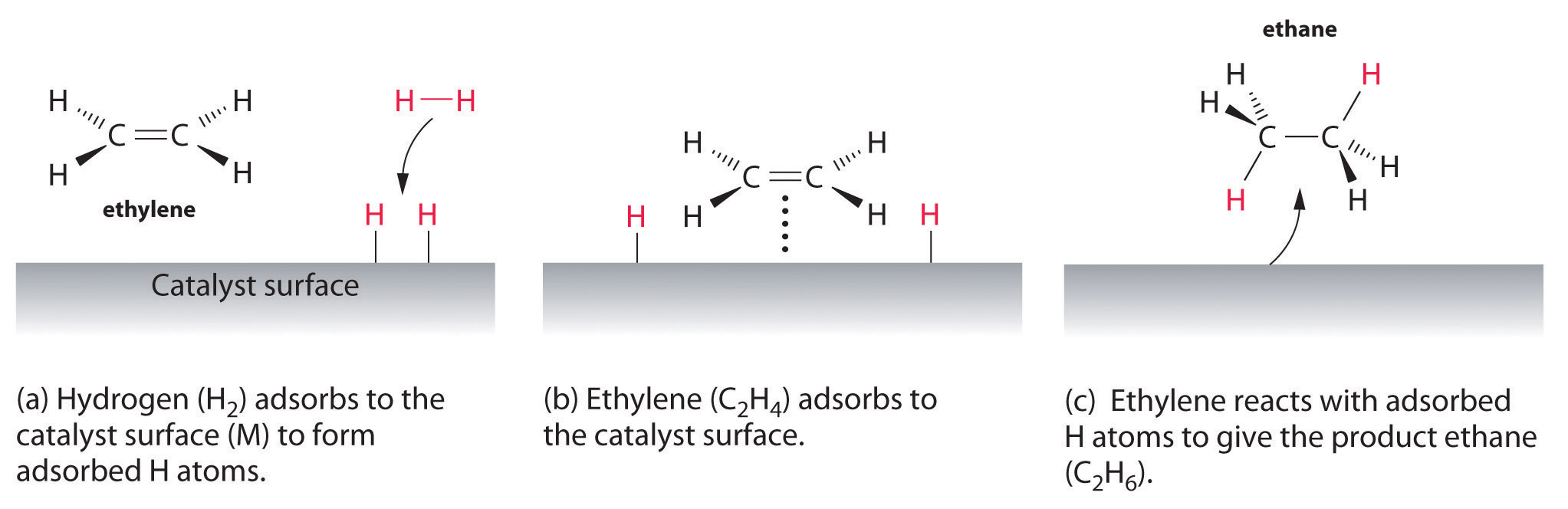
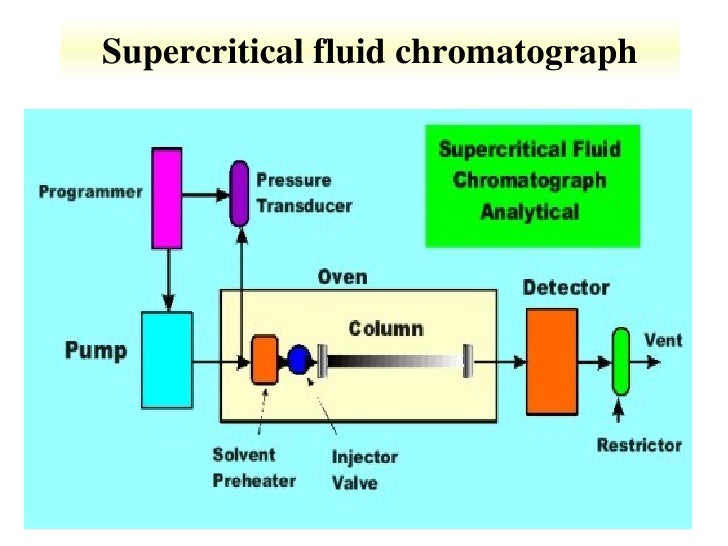
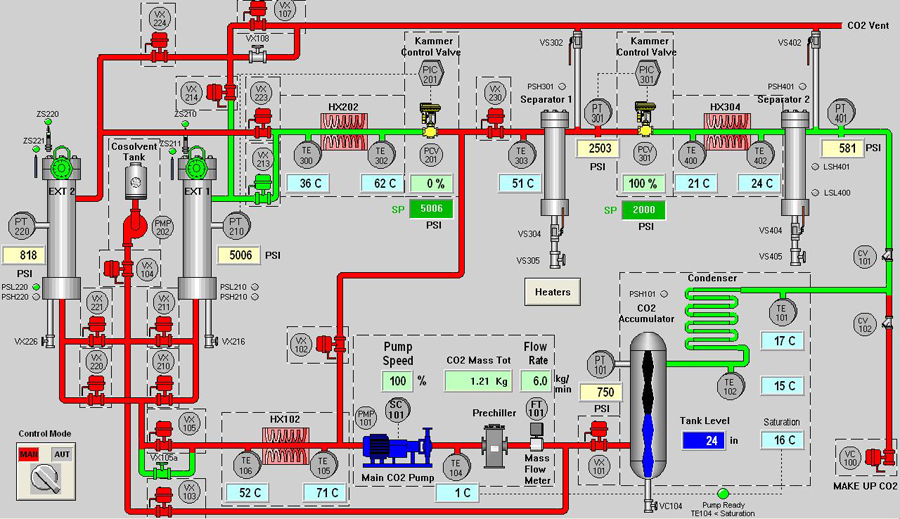
Schematic Diagram of sample Supercritical CO2 system
Table 1. Critical properties of various solvents (Reid et al., 1987)
| Solvent |
Molecular weight |
Critical temperature |
Critical pressure |
Critical density |
| g/mol |
K |
MPa (atm) |
g/cm3 |
| Carbon dioxide (CO2) |
44.01 |
304.1 |
7.38 (72.8) |
0.469 |
| Water (H2O) (acc. IAPWS) |
18.015 |
647.096 |
22.064 (217.755) |
0.322 |
| Methane (CH4) |
16.04 |
190.4 |
4.60 (45.4) |
0.162 |
| Ethane (C2H6) |
30.07 |
305.3 |
4.87 (48.1) |
0.203 |
| Propane (C3H8) |
44.09 |
369.8 |
4.25 (41.9) |
0.217 |
| Ethylene (C2H4) |
28.05 |
282.4 |
5.04 (49.7) |
0.215 |
| Propylene (C3H6) |
42.08 |
364.9 |
4.60 (45.4) |
0.232 |
| Methanol (CH3OH) |
32.04 |
512.6 |
8.09 (79.8) |
0.272 |
| Ethanol (C2H5OH) |
46.07 |
513.9 |
6.14 (60.6) |
0.276 |
| Acetone (C3H6O) |
58.08 |
508.1 |
4.70 (46.4) |
0.278 |
| Nitrous oxide (N2O) |
44.013 |
306.57 |
7.35 (72.5) |
0.452 |
Table 2 shows density, diffusivity and viscosity for typical liquids, gases and supercritical fluids.
Comparison of Gases, Supercritical Fluids and Liquids
|
Density (kg/m3) |
Viscosity (µPa∙s) |
Diffusivity (mm²/s) |
| Gases |
1 |
10 |
1–10 |
| Supercritical Fluids |
100–1000 |
50–100 |
0.01–0.1 |
| Liquids |
1000 |
500–1000 |
0.001 |
- F. Sahena, I. S. M. Zaidul, S. Jinap, A. A. Karim, K. A. Abbas, N. A. N. Norulaini and A. K. M. Omar, J. Food Eng., 2009, 95, 240–253
- D. J. Dixon and K. P. Jhonston, in Encyclopedia of Separation Technology, ed. D. M. Ruthven, John Wiley, 1997, 1544–1569
- P. Licence, J. Ke, M. Sokolova, S. K. Ross and M. Poliakoff, Green Chem., 2003, 5, 99–104
- X. Han and M. Poliakoff, Chem. Soc. Rev., 2012, 41, 1428–1436
- S. Marre, Y. Roig and C. Aymonier, J. Supercrit. Fluids, 2012, 66, 251–264
- F. Benito-Lopez, R. M. Tiggelaar, K. Salbut, J. Huskens, R. J. M. Egberink, D. N. Reinhoudt, H. J. G. E. Gardeniers and W. Verboom, Lab Chip, 2007, 7, 1345–1351
- R. Holliday, B. Y. M. Jong and J. W. Kolis, J. Supercrit. Fluids, 1998, 12, 255–260
- P. A. Hamley, T. Ilkenhans, J. M. Webster, E. García-Verdugo, E. Vernardou, M. J. Clarke, R. Auerbach, W. B. Thomas, K. Whiston and M. Poliakoff, Green Chem., 2002, 4, 235–238
- E. Pérez, J. Fraga-Dubreuil, E. García-Verdugo, P. A. Hamley, M. L. Thomas, C. Yan, W. B. Thomas, D. Housley, W. Partenheimer and M. Poliakoff, Green Chem., 2011, 13, 2397–2407




सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Linkedin

Join me on Facebook  FACEBOOK
FACEBOOK
Join me on twitter

 amcrasto@gmail.com
amcrasto@gmail.com

LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy
सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।