Tenofovir Disoproxil Fumarate
For full details see end of page

PAPER
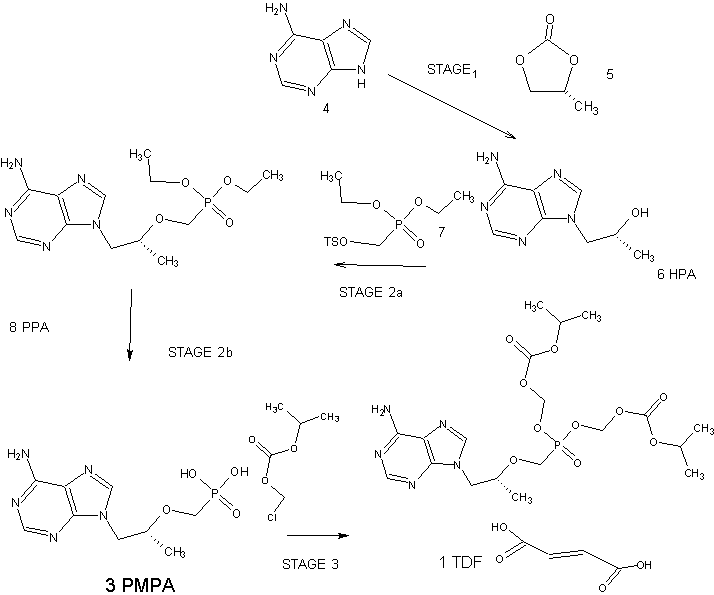
The current three-step manufacturing route for the preparation of tenofovir disoproxil fumarate (1) was assessed and optimized leading to a higher yielding, simpler, and greener process. Key improvements in the process route include the refinement of the second stage through the replacement of the problematic magnesium tert-butoxide (MTB) with a 1:1 ratio of a Grignard reagent and tert-butanol. The development of a virtually solvent-free approach and the establishment of a workup and purification protocol which allows the isolation of a pure diethyl phosphonate ester (8) was achieved
see………….http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00364
An Improved Process for the Preparation of Tenofovir Disoproxil Fumarate
† Department of Chemistry, Natural and Agricultural Sciences, University of Pretoria, 2 Lynnwood Road, Hatfield, 0002, Gauteng, South Africa
‡ Department of Engineering and Technology Management, University of Pretoria, Pretoria, South Africa
§ Pharmaceutical Manufacturing Technology Centre, University of Limerick, Limerick, V94 T9PX, Republic of Ireland
∥ iThemba Pharmaceuticals, Modderfontein, 1645, Gauteng South Africa
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.5b00364
Publication Date (Web): March 04, 2016
Copyright © 2016 American Chemical Society
-
Department of Chemistry
Pretoria, South Africa

Department of Chemistry, Natural and Agricultural Sciences, University of Pretoria, 2 Lynnwood Road, Hatfield, 0002, Gauteng, South Africa


///////
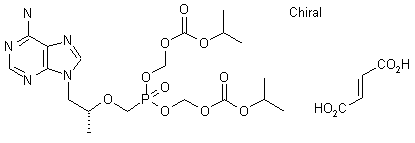
Tenofovir Disoproxil Fumarate
5-[[(1R)-2-(6-Amino-9H-purin-9-yl)-1-methylethoxy]methyl]-2,4,6,8-tetraoxa-5-phosphanonanedioic Acid 1,9-Bis(1-methylethyl) Ester 5-Oxide (2E)-2-Butenedioate; GS 4331-05; PMPA Prodrug; Tenofovir DF; Virea; Viread;
GILEAD-4331-300
201341-05-1 – free base, (Tenofovir Disoproxil
| CAS No.: |
202138-50-9 |
| Name: |
Tenofovir disoproxil fumarate |
| Molecular Structure: |
 |
| Formula: |
C19H30N5O10P.C4H4O4 |
| Molecular Weight: |
635.51 |
| Synonyms: |
TDF;PMPA prodrug;Tenofovir Disoproxil Fumarate [USAN];9-((R)-2-((Bis(((isopropoxycarbonyl)oxy)methoxy)phosphinyl)methoxy)propyl)adenine, fumarate;201341-05-1;Bis(NeopentylOC)PMPA;Viread;GS 4331-05 (*1:1 Fumarate salt*);Viread (*1:1 Fumarate salt*);Truvada;Tenofovir DF;[[(2R)-1-(6-aminopurin-9-yl)propan-2-yl]oxymethyl-(propan-2-yloxycarbonyloxymethoxy)phosphoryl]oxymethyl propan-2-yl carbonate; |
Usage
tyrosinase inhibitor used for skin lightening and anti-melasma |
|
Usage
An acyclic phosphonate nucleotide analog and selective HIV-1 RT inhibitor |
|
Usage
Acyclic phosphonate nucleotide analogue; reverse transcriptase inhibitor. Used as an anti-HIV agent. Antiviral. |
|
Tenofovir disoproxil is an antiretroviral medication used to prevent and treat HIV/AIDS and to treat chronic hepatitis B.[1] The active substance is tenofovir, while tenofovir disoproxil is a prodrug that is used because of its better absorption in the gut.
The drug is on the World Health Organization’s List of Essential Medicines, the most important medications needed in a basic health system.[2] It is marketed by Gilead Sciences under the trade name Viread (as the fumarate, TDF).[3] As of 2015 the cost for a typical month of medication in the United States is more than 200 USD.[4]

Medical uses
- HIV-1 infection: Tenofovir is indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and pediatric patients 2 years of age and older.[5] This indication is based on analyses of plasma HIV-1 RNA levels and CD4 cell counts in controlled studies of tenofovir in treatment-naive and treatment-experienced adults.
- Tenofovir is indicated for the treatment of chronic hepatitis B in adults and pediatric patients 12 years of age and older.[5][6]
HIV risk reduction
A Cochrane review examined the use of tenofovir for prevention of HIV before exposure. It found that both tenofovir alone and the tenofovir/emtricitabine combination decreased the risk of contracting HIV.[7]
The U. S. Centers for Disease Control and Prevention (CDC) conducted a study in partnership with the Thailand Ministry of Public Health to ascertain the effectiveness of providing people who inject drugs illicitly with daily doses of the antiretroviral drug tenofovir as a prevention measure. The results of the study were released in mid-June 2013 and revealed a 48.9%-reduced incidence of the virus among the group of subjects who received the drug, in comparison to the control group who received a placebo. The principal investigator of the study stated: “We now know that pre-exposure prophylaxis can be a potentially vital option for HIV prevention in people at very high risk for infection, whether through sexual transmission or injecting drug use.”[8]
Adverse effects
The most common side effects associated with tenofovir include nausea, vomiting, diarrhea, and asthenia. Less frequent side effects include hepatotoxicity, abdominal pain, and flatulence.[9] Tenofovir has also been implicated in causing renal toxicity, particularly at elevated concentrations.[10]
Tenofovir can cause acute renal failure, Fanconi syndrome, proteinuria, or tubular necrosis.[citation needed] These side effects are due to accumulation of the drug in proximal tubules.[citation needed] Tenofovir can interact with didanosine by increasing didanosine’s concentration.[citation needed] It also decreases the concentration of atazanavir sulfate.[citation needed]
Mechanism of action
Tenofovir is a defective adenosine nucleotide that selectively interferes with the action of reverse transcriptase, but only weakly interferes with mammalian DNA polymerases α, β, and mitochondrial DNA polymerase γ.[11] Tenofovir prevents the formation of the 5′ to 3′ phosphodiester linkage essential for DNA chain elongation. A phosphodiester bond cannot be formed because the tenofovir molecule lacks an —OH group on the 3′ carbon of its deoxyribose sugar.[11] Once incorporated into a growing DNA strand, tenofovir causes premature termination of DNA transcription. The drug is classified as a nucleotide analogue reverse transcriptase inhibitor (NRTI), that inhibits reverse transcriptase.[11] Reverse transcriptase is a crucial viral enzyme in retroviruses such as human immunodeficiency virus (HIV) and in hepatitis B virus infections.[5]
History
Tenofovir was initially synthesized by Antonín Holý at the Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic in Prague. The patent[12] filed by Holý in 1984 makes no mention of the potential use of the compound for the treatment of HIV infection, which had only been discovered one year earlier.
In 1985, De Clercq and Holý described the activity of PMPA against HIV in cell culture.[13] Shortly thereafter, a collaboration with the biotechnology company Gilead Sciences led to the investigation of PMPA’s potential as a treatment for HIV infected patients. In 1997 researchers from Gilead and the University of California, San Francisco demonstrated that tenofovir exhibits anti-HIV effects in humans when dosed by subcutaneous injection.[14]
The initial form of tenofovir used in these studies had limited potential for widespread use because it was not absorbed when administered orally. A medicinal chemistry team at Gilead developed a modified version of tenofovir, tenofovir disoproxil.[15] This version of tenofovir is often referred to simply as “tenofovir”. In this version of the drug, the two negative charges of the tenofovir phosphonic acid group are masked, thus enhancing oral absorption.
Tenofovir disoproxil was approved by the U.S. FDA on October 26, 2001, for the treatment of HIV, and on August 11, 2008, for the treatment of chronic hepatitis B.[16][17]
Drug forms
Tenofovir disoproxil is a prodrug form of tenofovir. It is also marketed under the brand name Reviro by Dr. Reddy’s Laboratories. Tenofovir is also available in a fixed-dose combination with emtricitabine in a product with the brand name Truvada for once-a-day dosing. Efavirenz/emtricitabine/tenofovir disoproxil (brand name Atripla) — a fixed-dose triple combination of tenofovir, emtricitabine, and efavirenz, was approved by the FDA on 12 July 2006 and is now available, providing a single daily dose for the treatment of HIV.
Therapeutic drug monitoring
Tenofovir may be measured in plasma by liquid chromatography. Such testing is useful for monitoring therapy and to prevent drug accumulation and toxicity in people with kidney or liver problems.[18][19][20]
PATENT
http://www.google.com/patents/EP2545063A2?cl=en
Tenofovir Disoproxil is chemically known as 9-[-2-(R)-[[bis [[(isopropoxycarbonyl) oxy]methoxy] phosphinoyl]methoxy]propyl]-adenine, having the following structural formula-I.
Formula-I
Tenofovir is a highly potent antiviral agent, particularly for the therapy or prophylaxis of retroviral infections and belongs to a class of drugs called Nucleotide Reverse Transcriptase Inhibitors (NRTI) which blocks reverse transcriptase an enzyme crucial to viral production in HIV-infected people.
Tenofovir Disoproxil and its pharmaceutically acceptable salts were first disclosed in US 5,922,695. This patent discloses the preparation of Tenofovir Disoproxil by the esterification of Tenofovir with chloromethyl isopropyl carbonate using l-methyl-2- pyrrolidinone and triethylamine. In this patent Tenofovir Disoproxil is converted into its Fumarate salt without isolation. PCT Publication WO 2008007392 discloses process for the preparation of Tenofovir Disoproxil fumarate, wherein the isolated crystalline Tenofovir Disoproxil is converted into fumarate salt.
Tenofovir Disoproxil processes in the prior art are similar to process disclosed in product patent US 5,922,695. According to the prior art processes, Tenofovir Disoproxil fumarate obtained is having low yields and also show the presence of impurities such as dimers.
scheme- 1.
Tenofovir disoproxil chloromethyl isopropyl carbonate
Tenofovir disoproxil fumarate
Example 1 : Process for the preparation of Tenofovir Disoproxil fumarate
Toluene (500 ml) was added to the Tenofovir (100 gm) and stirred at room temperature. To this triethylamine (66.31 gm) was added, temperature was raised to 90° C and water was collected by azeotropic distillation at 110°C. Toluene was completely distilled under vacuum at same temperature. The reaction mixture was cooled to room temperature and to this a mixture of N-methyl pyrrolidine (300 gm), triethylamine (66.31 gm), Tetrabutyl ammonium bromide (52.8 gm) and trimethyl silyl chloride (17.8 gm) were added. The above reaction mixture was heated to 50-55 °C and was added slowly chloromethyl. isopropyl carbonate (CMIC) and maintained the reaction mixture at 50-55°C for 5 hrs. (Qualitative HPLC analysis shows about 85% product formation). The above reaction mixture was cooled to room temperature and filtered. The filtrate was added to DM water at 5-10°C and extract with dichloromethane. The combined dichloromethane layer was concentrated under vacuum and the crude was Co-distilled with cyclohexane and this crude was taken into isopropyl alcohol (1000 ml). To this fumaric acid (38 gm) was added and temperature was raised to 50° C. The reaction mixture was filtered and filtrate was cooled to 5-10° C. The obtained solid was filtered and washed with isopropyl alcohol. The compound was dried under vacuum to yield Tenofovir Disoproxil fumarate (140 gm).
Example-2 : Preparation of Tenofovir
N-methyl-2-pyrrolidone (25 gm) was taken along with toluene (150 gm) into a reaction vessel. l-(6-amino-purin-9-yl)-propan-2-ol (100 gm); toluene-4-sulfonic acid diethoxy phosphoryl methyl ester (200 gm) and magnesium ter-butoxide (71.2 gm) were also taken at’ 25-35°C. Temperature was raised to 74-75 °C and maintained for 5-6hrs. After completion of reaction, acetic acid (60 gm) was added and maintained for 1 hr. Later aq.HBr (332 gm) was taken and heated to 90-95 °C. After reaction completion, salts were filtered and filtrate was subjected to washings with water and extracted into methylene dichloride. Later pH was adjusted using CS lye below 10 °C. Tenofovir product was isolated using acetone.
Yield: 110 gm.
Example 3 : Preparation of Tenofovir disoproxil
(R)-9-[2-(phosphonomethoxy)propyl]adenine (25 gm), triethyl amine (25 ml) and cyclohexane (200 ml) were combined and heated to remove water and the solvent was distilled off under vacuum. The reaction mass was cooled to room temperature N-methyl pyrrolidinone (55 ml), triethyl amine (25 ml) and tetra butyl ammonium bromide(54 gms) were added to the reaction mixture. The reaction mass was heated to 50-60°C and chloromethyl isopropyl carbonate (65 gm) was added and maintained for 4-8 hrs at 50- 60°C and then cooled to 0°C. The reaction mass was diluted with chilled water or ice and precipitated solid product was filtered. The mother liquor was extracted with methylene chloride (150 ml). The methylene chloride layer was washed with water (200 ml). The filtered solid and the methylene chloride layer were combined and washed with water and the solvent was distilled under vacuum. Ethyl acetate was charged to the precipitated solid. The reaction mass was then cooled to 0-5 °C and maintained for 6 hrs. The solid was filtered and dried to produce Tenofovir disoproxil (45 gm).
CLIPS
The reaction of chloromethyl chloroformate (I) with isopropyl alcohol (II) by means of pyridine or triethylamine in ether gives the mixed carbonate (III), which is then condensed with (R)-PMPA (IV) by means of diisopropyl ethyl-amine in DMF.
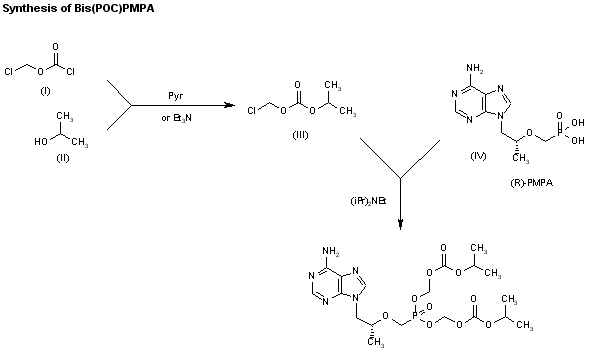
CLIP 2
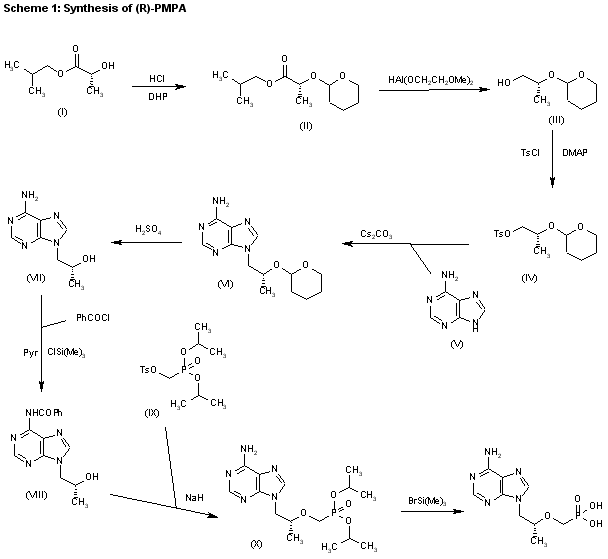
1) The protection of isobutyl D-(+)-lactate (I) with dihydropyran (DHP)/HCl in DMF gives the tetrahydropyranyloxy derivative (II), which is reduced with bis(2-methoxyethoxy)aluminum hydride in refluxing ether/ toluene yielding 2(R)-(tetrahydropyranyloxy)-1-propanol (III). The tosylation of (III) with tosyl chloride as usual affords the expected tosylate (VI), which is condensed with adenine (V) by means of Cs2CO3 in hot DMF, affording 9-[2(R)-(tetrahydropyranyloxy)propyl]adenine (VI). The deprotection of (VI) with sulfuric acid affords 9-[2(R)-hydroxypropyl]adenine (VII), which is N-benzoylated with benzoyl chloride/chlorotrimethylsilane in pyridine to give the benzamide (VIII), which is condensed with tosyl-oxymethylphosphonic acid diisopropyl ester (IX) by means of NaH in DMF to yield 9-[2(R)-(diisopropoxyphosphorylmethoxy)propyl]adenine (X). Finally, this compound is hydrolyzed by means of bromotrimethylsilane in acetonotrile.
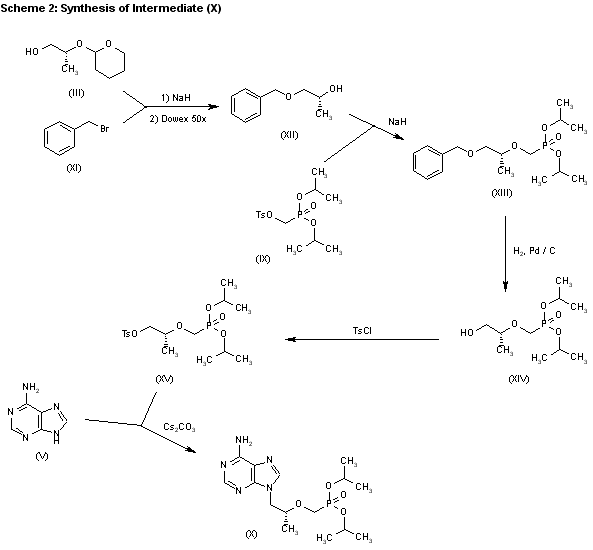
2) The reaction of the previously described (R)-2-(2-tetrahydropyranyloxy)-1-propanol (III) with benzyl bromide (XI) by means of NaH in DMF, followed by a treatment with Dowex 50X, gives 1-benzyloxy-2(R)-propanol (XII), which is condensed with tosyloxymethylphosphonic acid diisopropyl ester (IX) by means of NaH in THF, yielding 2-benzyloxy-1(R)-methylethoxymethylphosphonic acid diisopropyl ester (XIII). The hydrogenolysis of (XIII) over Pd/C in methanol affords 2-hydroxy-1(R)-methylethoxymethylphosphonic acid diisopropyl ester (XIV), which is tosylated with tosyl chloride/dimethyl-aminopyridine in pyridine to give the expected tosylate (XV). The condensation of (XV) with adenine (VI) by means of Cs2CO3 in hot DMF yields 9-[2(R)-(diisopropoxyphosphorylmethoxy)propyl]adenine (X), which is finally hydrolyzed as before.
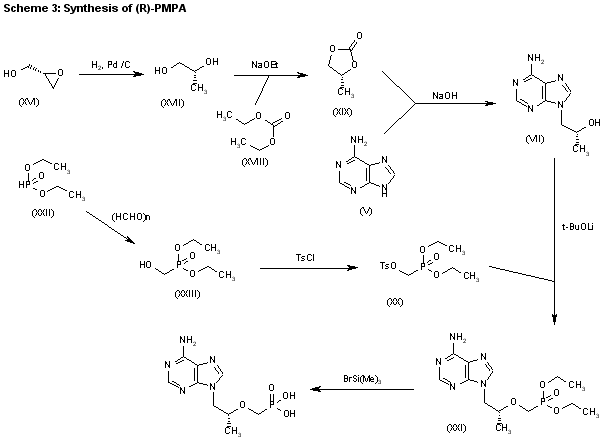
3) The catalytic hydrogenation of (S)-glycidol (XVI) over Pd/C gives the (R)-1,2-propanediol (XVII), which is esterified with diethyl carbonate (XVIII)/NaOEt, yielding the cyclic carbonate (XIX). The reaction of (XIX) with adenine (V) by means of NaOH in DMF affords 9-[2(R)-hydroxypropyl]adenine (VII), which is condensed with tosyloxymethylphosphonic acid diethyl ester (XX) by means of lithium tert-butoxide in THF, giving 9-[2(R)-(diethoxyphosphorylmethoxy)propyl]adenine (XXI). Finally, this compound is hydrolyzed with bromotrimethylsilane as before. Compound (XX) is obtained by reaction of diethyl phosphite (XXII) with paraformaldehyde, yielding hydroxy- methylphosphonic acid diethyl ester (XXIII), which is finally tosylated as usual.
References
- 1 “Viread”. The American Society of Health-System Pharmacists. Retrieved 31 July 2015.
- 2
- “WHO Model List of EssentialMedicines” (PDF). World Health Organization. October 2013. Retrieved 22 April 2014.
- 3
- Emau P, Jiang Y, Agy MB, et al. (2006). “Post-exposure prophylaxis for SIV revisited: Animal model for HIV infection”. AIDS Res Ther 3: 29. doi:10.1186/1742-6405-3-29. PMC 1687192. PMID 17132170.
- 4
- Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 66. ISBN 9781284057560.
- 5
- Gilead Sciences, Inc. Prescribing Information. Revised: November 2012.
- 6
- Guidelines for the prevention, care and treatment of persons with chronic hepatitis B infection, WHO, Publication details:Pages: 166 Publication date: March 2015 Languages: English ISBN 978 92 4 154905 9
- 7
- Okwundu CI, Uthman OA, Okoromah CAN (2012). “Antiretroviral pre-exposure prophylaxis (PrEP) for preventing HIV in high-risk individuals”. Cochrane Database Syst Rev 7 (7): CD007189. doi:10.1002/14651858.CD007189.pub3. PMID 22786505.
- 8
- Emma Bourke (14 June 2013). “Preventive drug could reduce HIV transmission among injecting drug users”. The Conversation Australia. The Conversation Media Group. Retrieved 17 June 2013.
- 9
- USPDI. Thompson. 2005. pp. 2741–2.
- 10
- “Viread Prescribing Guidelines” (PDF). U.S. Food and Drug Administration. March 2006. Archived from the original (PDF) on 2007-09-30. Retrieved 2007-02-12.
- 11
- Drugbank: Tenofovir
- 12
- “Patent US4808716 – 9-(phosponylmethoxyalkyl) adenines, the method of preparation and … – Google Patents”.
- 13
- A US 4724233 A, De Clercq, Erik; Antonin Holy & Ivan Rosenberg, “Therapeutical application of phosphonylmethoxyalkyl adenines”, published 1985-04-25
- 14
- Deeks SG, Barditch-Crovo P, Lietman PS, et al. (September 1998). “Safety, pharmacokinetics, and antiretroviral activity of intravenous 9-[2-(R)-(Phosphonomethoxy)propyl]adenine, a novel anti-human immunodeficiency virus (HIV) therapy, in HIV-infected adults”. Antimicrob. Agents Chemother. 42 (9): 2380–4. PMC 105837. PMID 9736567.
- 15
- “Patent US5977089 – Antiviral phosphonomethoxy nucleotide analogs having increased oral … – Google Patents”.
- 16
- FDA letter of approval (regarding treatment of hepatitis B)
- 17
- FDA Clears Viread for Hepatitis B
- 18
- Delahunty T, Bushman L, Robbins B, Fletcher CV (2009). “The simultaneous assay of tenofovir and emtricitabine in plasma using LC/MS/MS and isotopically labeled internal standards”. J. Chrom. B 877 (20–21): 1907–1914. doi:10.1016/j.jchromb.2009.05.029.
- 19
- Kearney BP, Yale K, Shah J, Zhong L, Flaherty JF (2006). “Pharmacokinetics and dosing recommendations of tenofovir disoproxil fumarate in hepatic or renal impairment”. Clin. Pharmacokinet. 45 (11): 1115–24. doi:10.2165/00003088-200645110-00005. PMID 17048975.
- 20
- R. Baselt, Disposition of Toxic Drugs and Chemicals in Man, 8th edition, Biomedical Publications, Foster City, California, 2008, pp. 1490–1492.
External links
| WO2008007392A2 |
Jul 11, 2007 |
Jan 17, 2008 |
Matrix Lab Ltd |
Process for the preparation of tenofovir |
| US5922695 |
Jul 25, 1997 |
Jul 13, 1999 |
Gilead Sciences, Inc. |
Antiviral phosphonomethyoxy nucleotide analogs having increased oral bioavarilability |
| WO2015051874A1 |
Sep 22, 2014 |
Apr 16, 2015 |
Zentiva, K.S. |
An improved process for the preparation of tenofovir disoproxil and pharmaceutically acceptable salts thereof |
| CN103360425A * |
Apr 1, 2012 |
Oct 23, 2013 |
安徽贝克联合制药有限公司 |
Synthesis method of tenofovir disoproxil and fumarate thereof |
| CN103374038A * |
Apr 11, 2012 |
Oct 30, 2013 |
广州白云山制药股份有限公司广州白云山制药总厂 |
Preparation method of antiviral medicine |
| CN103848868A * |
Dec 4, 2012 |
Jun 11, 2014 |
蚌埠丰原涂山制药有限公司 |
Method for preparing tenofovir |
| CN103848869A * |
Dec 4, 2012 |
Jun 11, 2014 |
上海医药工业研究院 |
Method for preparing tenofovir |
| CN103980319A * |
Apr 24, 2014 |
Aug 13, 2014 |
浙江外国语学院 |
Preparation method of tenofovir |
| CN103980319B * |
Apr 24, 2014 |
Dec 2, 2015 |
浙江外国语学院 |
一种泰诺福韦的制备方法 |
| EP2860185A1 |
Oct 9, 2013 |
Apr 15, 2015 |
Zentiva, k.s. |
An improved process for the preparation of Tenofovir disoproxil and pharmaceutically acceptable salts thereof |
The chemical name of tenofovir disoproxil fumarate is 9-[(R)-2[[bis[[(isopropoxycarbonyl)oxy]methoxy]phosphinyl]methoxy]propyl]adenine fumarate (1:1). It has a molecular formula of C19H30N5O10P • C4H4O4 and a molecular weight of 635.52. It has the following structural formula:
Tenofovir disoproxil fumarate is a white to off-white crystalline powder with a solubility of 13.4 mg/mL in distilled water at 25 °C. It has an octanol/phosphate buffer (pH 6.5) partition coefficient (log p) of 1.25 at 25 °C.
VIREAD is available as tablets or as an oral powder.
VIREAD tablets are for oral administration in strengths of 150, 200, 250, and 300 mg of tenofovir disoproxil fumarate, which are equivalent to 123, 163, 204 and 245 mg of tenofovir disoproxil, respectively. Each tablet contains the following inactive ingredients: croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, and pregelatinized starch. The 300 mg tablets are coated with Opadry II Y-3010671-A, which contains FD&C blue #2 aluminum lake, hypromellose 2910, lactose monohydrate, titanium dioxide, and triacetin. The 150, 200, and 250 mg tablets are coated with Opadry II 32K-18425, which contains hypromellose 2910, lactose monohydrate, titanium dioxide, and triacetin.
VIREAD oral powder is available for oral administration as white, taste-masked, coated granules containing 40 mg of tenofovir disoproxil fumarate per gram of oral powder, which is equivalent to 33 mg of tenofovir disoproxil. The oral powder contains the following inactive ingredients: mannitol, hydroxypropyl cellulose, ethylcellulose, and silicon dioxide.
///////