












Trimyristin
Trimyristin is an ester with the chemical formula C45H86O6. It is a saturated fat which is the triglyceride of myristic acid. Trimyristin is a white to yellowish-gray solid that is insoluble in water, but soluble in ethanol, benzene, chloroform, dichloromethane, and ether.
| Name | Trimyristin |
| Synonyms | Glycerol trimyristate |
| Name in Chemical Abstracts | Tetradecanoic acid, 1,2,3-propanetriyl ester |
| CAS No | 555-45-3 |
| EINECS No | 209-099-7 |
| Molecular formula | C45H86O6 |
| Molecular mass | 723.18 |
| SMILES code | CCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCC)COC(=O)CCCCCCCCCCCCC |
 |
|
|

Occurrence
Trimyristin is found naturally in many vegetable fats and oils.
Isolation from nutmeg

The isolation of trimyristin from powdered nutmeg is a common introductory-level college organic chemistry experiment. It is an uncommonly simple natural product extraction because nutmeg oil generally consists of over eighty percent trimyristin. Trimyristin makes up between 20-25% of the overall mass of dried, ground nutmeg. Separation is generally carried out by steam distillation and purification uses extraction from ether followed by distillation or rotary evaporation to remove the volatile solvent. The extraction of trimyristin can also be done with diethyl ether at room temperature, due to its high solubility in the ether. The experiment is frequently included in curricula, both for its relative ease and to provide instruction in these techniques.
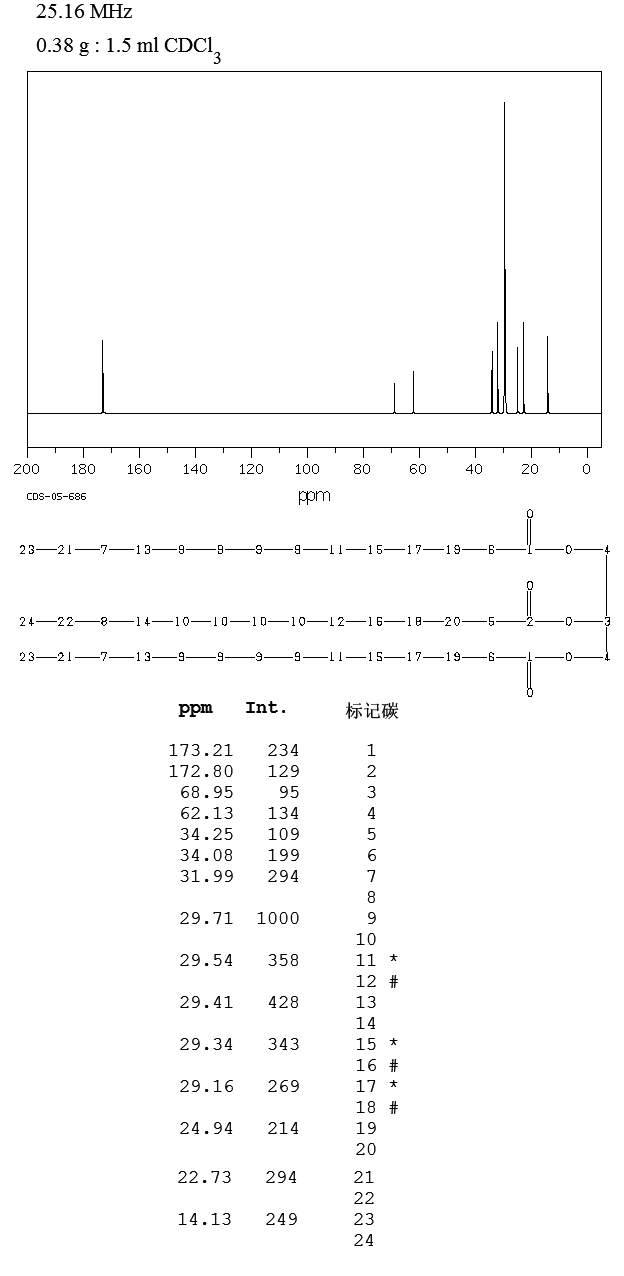
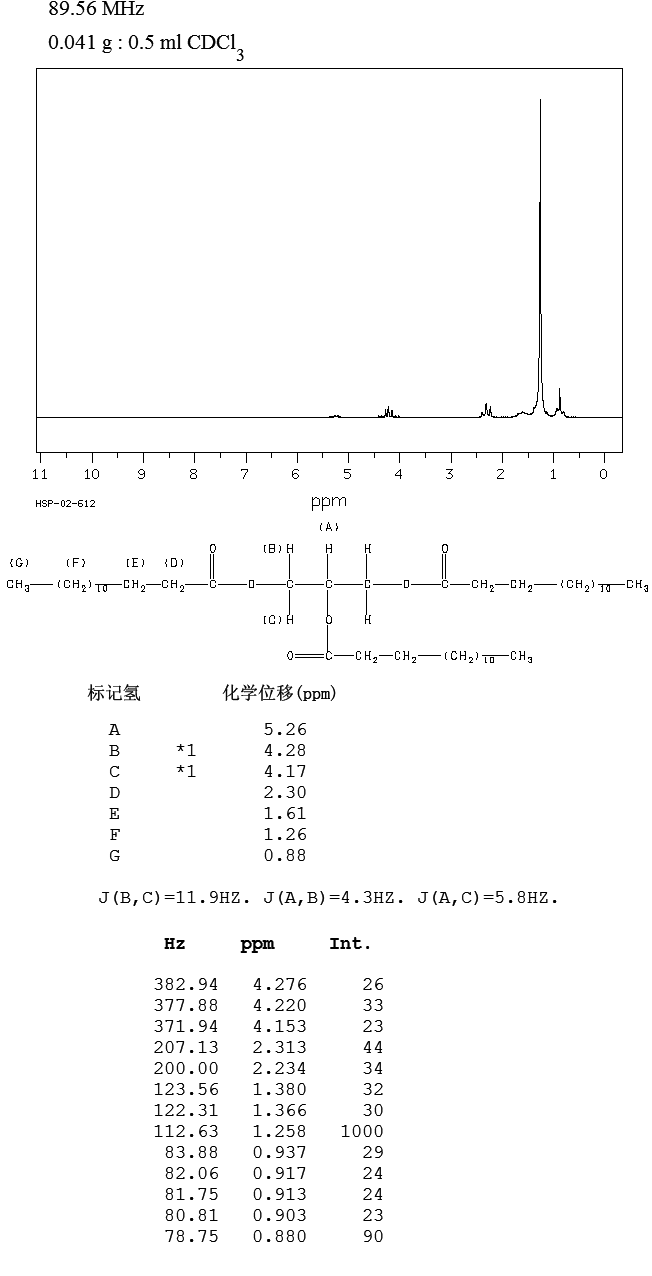
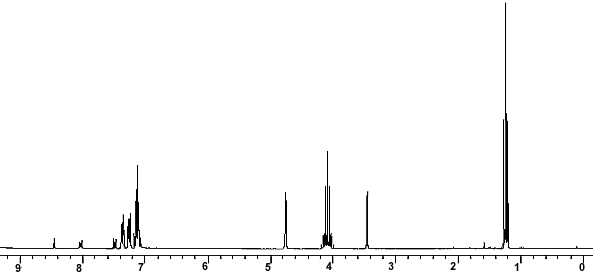
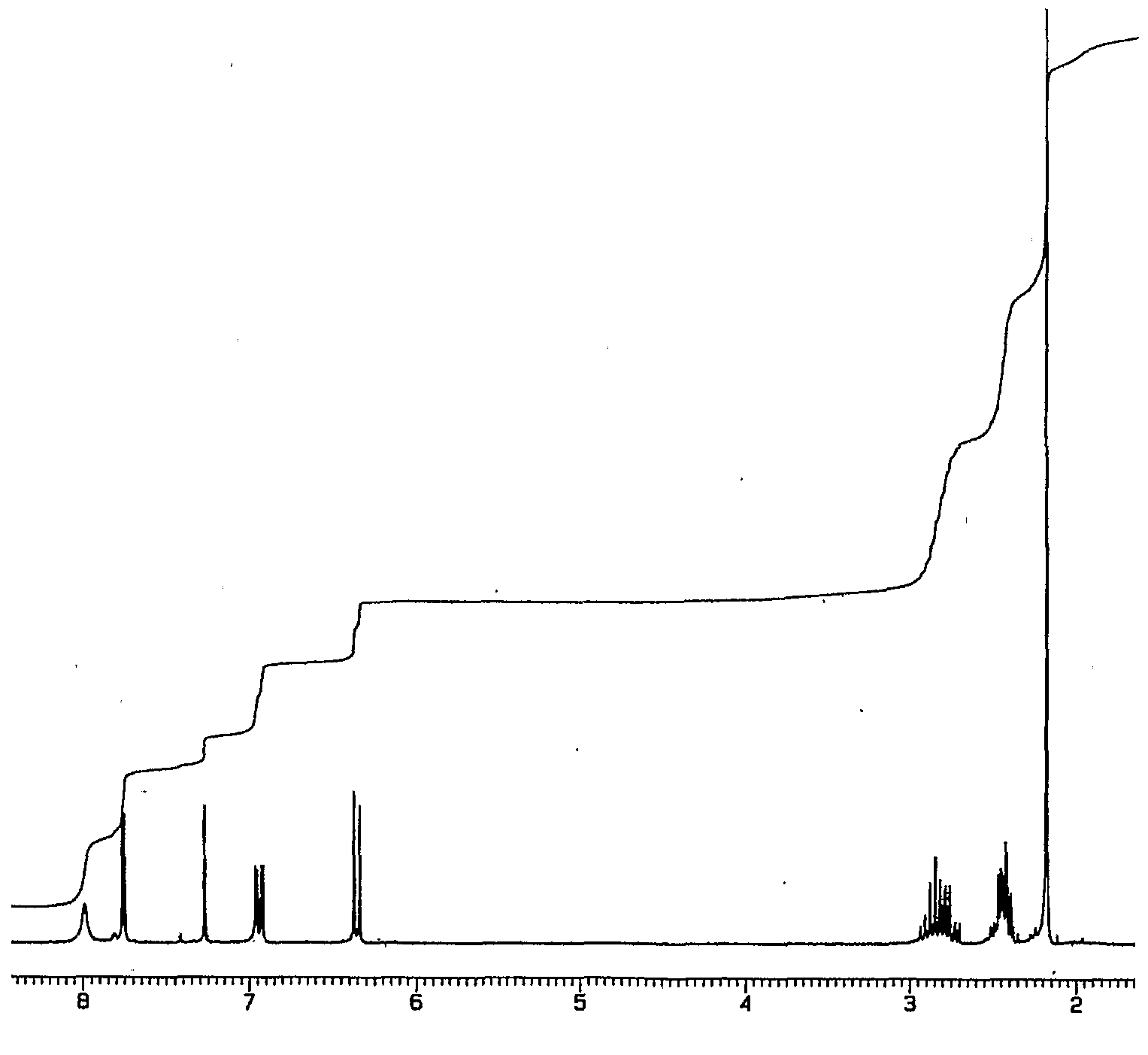
1H-NMR

| 1H-NMR: Trimyristin | |||
| 300 MHz, CDCl3 | |||
| delta [ppm] | mult. | atoms | assignment |
| 0.90 | m | 9 H | 14-H (CH3) |
| 1.2-1.4 | m | 60 H | 4-13-H (CH2) |
| 1.5-1.7 | m | 6 H | 3-H |
| 2.33 | m | 6 H | 2-H |
| 4.16 | dd | 2 H | glycerol-C1-Ha |
| 4.31 | dd | 2 H | glycerol-C1-Hb |
| 5.28 | m | 1 H | glycerol-C2-H |
| 7.26 | CHCl3 | ||
| 2.11 | acetone (impurity) | ||
Isolation of trimyristin from nutmeg |
| Reaction type: | isolation of natural products |
| Substance classes: | carboxylic acid ester, triglyceride, natural product |
| Techniques: | extracting with Soxhlet extractor, evaporating with rotary evaporator, recrystallizing, filtering, heating under reflux, heating with oil bath, stirring with magnetic stir bar |
| Degree of difficulty: | Easy |
| Batch scale: | 25 g | Nutmeg |
Reaction……….http://kriemhild.uft.uni-bremen.de/nop/en/instructions/pdf/1021_en.pdfThe reaction apparatus consists of a 250 mL round-bottom flask with a magnetic stir bar and a 100 mL soxhlet extraction unit with a reflux condenser. 25 g of finely ground nutmeg are placed into the extraction sleeve and covered with a little glass wool. 150 mL tert-butyl methyl ether are placed into the flask and whilst stirring, the solvent is heated to reflux until the solvent leaving the extraction sleeve is colourless (approximately 5 hours). Work upThe solvent is evaporated with a final pressure of 20 hPa. The flask containing the residue is cooled in an ice bath or the refrigerator until the contents has crystallized to a thick slurry. Crude product yield: 12 g; The crude product is recrystallized from the minimum amount of ethanol. Prior to filtering the crystals, the flask is placed into the refrigerator for at least 30 minutes. The crystalline slurry is filtered and the product is dried in an evacuated desiccator over silica gel. Should the crystals not be colourless after the first recrystallization, a second recrystallization is carried out. Yield: 6.5 g; melting point 54-55 °C; Duration of the experimentWithout recrystallization 6 hours Where can I stop the experiment?Before and after the evaporation of the solvent RecyclingThe evaporated tert-butyl methyl ether and the evaporated ethanol from the mother liquor are collected and redistilled. |
Suggestions for waste disposal |
||||||
|
Operating scheme

Substances required
| Educts | Amount | Risk | Safety | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Nutmeg |
|
25 g | R | S | ||||||
| Solvents | Amount | Risk | Safety | |||||||
| Ethanol |
|
~ 150 mL | R 11 | S 2-7-16 | ||||||
| tert-Butyl methyl ether |
|
150 mL | R 11-38 | S 2-9-16-24 | ||||||
| Others | Amount | Risk | Safety | |||||||
| Iodine |
|
0.1 g | R 20/21-50 | S 2-23.2-23.4-25-61 | ||||||
| Solvents for analysis | Amount | Risk | Safety | |||||||
| Cyclohexane |
|
? | R 11-38-50/53-65-67 | S 2-9-16-33-60-61-62 | ||||||
| Acetic acid ethyl ester |
|
? | R 11-36-66-67 | S 2-16-26-33 |
Substances produced
| Products | Amount | Risk | Safety | ||
|---|---|---|---|---|---|
| Trimyristin |
|
6.5 g | R | S |
Equipment
 |
round bottom flask 250 mL |  |
Soxhlet extractor 100 mL | |
 |
glass wool |  |
extraction cone | |
 |
heatable magnetic stirrer with magnetic stir bar |  |
oil bath | |
 |
reflux condenser |  |
rotary evaporator | |
 |
ice bath |  |
exsiccator with drying agent | |
 |
suction filter |  |
suction flask |
Simple evaluation indices
| Atom economy | not defined | ||
| Yield | not defined | ||
| Target product mass | 6.5 | g | |
| Sum of input masses | 250 | g | |
| Mass efficiency | 26 | mg/g | |
| Mass index | 39 | g input / g product | |
| E factor | 38 | g waste / g product | |
| Energy input | 1500 | kJ | |
| Energy efficiency | 4.3 | mg/kJ |
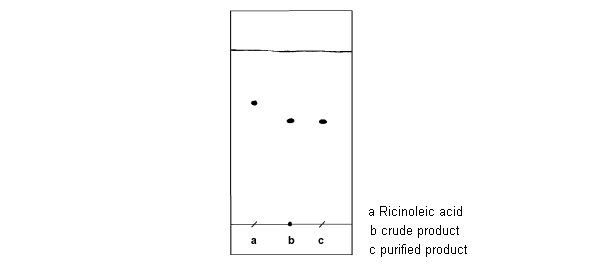
Chromatogram

| TLC: crude product | |
| TLC layer | Polygram SilG/UV precoated TLC layer; 0.2 mm; silica gel; Macherey & Nagel |
| mobile phase | cyclohexane / EtOAc = 95 : 5 |
| staining reagent | Vaughn’s reagent or iodine vapor |
| Rf (product) | 0.51 |
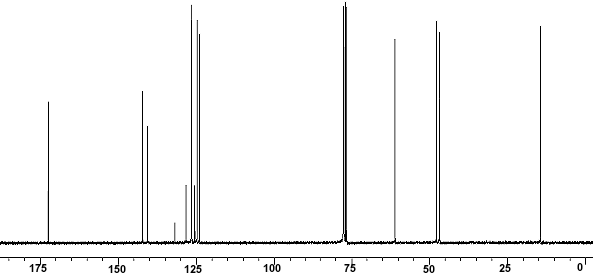
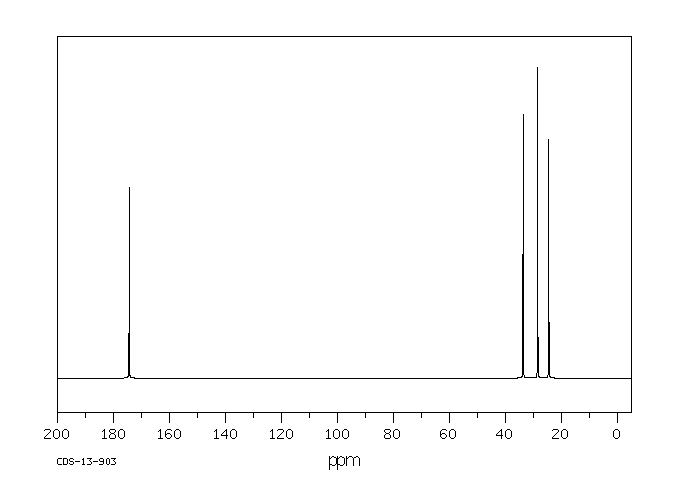
13C-NMR

| 13C-NMR: Trimyristin | |||
| 300 MHz, CDCl3 | |||
| delta [ppm] | assignment | ||
| 14.08 | C14 | ||
| 22.66 | C13 | ||
| 24.85-24.89 | C3, C17 | ||
| 29.06-31.90 | C4-C12 | ||
| 34.04-34.2 | C2 | ||
| 62.08 | glycerol-C1 | ||
| 68.85 | glycerol-C2 | ||
| 172.85 | C15 | ||
| 173.26 | C1 | ||
| 76.5-77.5 | CDCl3 | ||
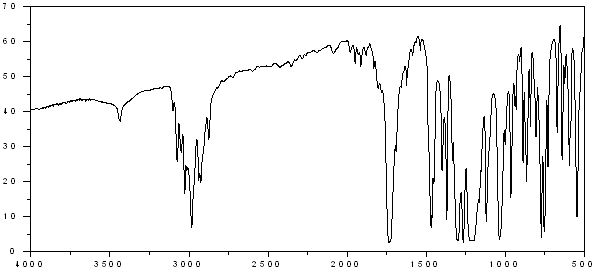
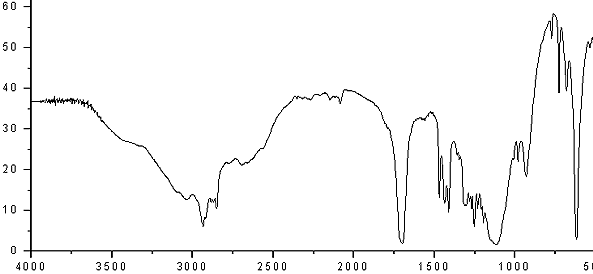
IR

| IR: Trimyristin | |||
| [KBr, T%, cm-1] | |||
| [cm-1] | assignment | ||
| 2950-2850 | aliph. C-H valence | ||
| 1730 | C=O valence, ester | ||
 |
|
 |
|
 |
|
| Names | |
|---|---|
| IUPAC name
1,3-Di(tetradecanoyloxy)propan-2-yl tetradecanoate
|
|
| Other names | |
| Identifiers | |
| 555-45-3 |
|
| ChemSpider | 10675 |
| EC number | 209-099-7 |
| Jmol-3D images | Image |
| PubChem | 11148 |
| UNII | 18L31PSR28 |
| Properties | |
| C45H86O6 | |
| Molar mass | 723.18 g·mol−1 |
| Appearance | White-yellowish gray solid |
| Odor | Nutmeg-like |
| Density | 0.862 g/cm3 (20 °C)[4] 0.8848 g/cm3 (60 °C)[2] |
| Melting point | 56–57 °C (133–135 °F; 329–330 K) |
| Boiling point | 311 °C (592 °F; 584 K) |
| Solubility | Slighty soluble in alcohol, ligroin Soluble in (C2H5)2O, acetone, C6H6,[2] CH2Cl2, CHCl3 |
|
Refractive index (nD)
|
1.4428 (60 °C)[2] |
| Structure | |
| Triclinic (β-form)[3] | |
| P1 (β-form)[3] | |
|
a = 12.0626 Å, b = 41.714 Å, c = 5.4588 Å (β-form)[3]
α = 73.888°, β = 100.408°, γ = 118.274°
|
|
| Thermochemistry | |
| 1013.6 J/mol·K (β-form, 261.9 K) 1555.2 J/mol·K (331.5 K)[5][6] |
|
|
Std molar
entropy (S |
1246 J/mol·K (liquid)[6] |
|
Std enthalpy of
formation (ΔfH |
−2355 kJ/mol[6] |
|
Std enthalpy of
combustion (ΔcH |
27643.7 kJ/mol[6] |
| Hazards | |
| NFPA 704 | |
| Flash point | > 110 °C (230 °F; 383 K)[7] |
| 421.1 °C (790.0 °F; 694.2 K)[7] | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|

-
References
- Merck Index, 11th Edition, 9638.
- Lide, David R., ed. (2009). CRC Handbook of Chemistry and Physics (90th ed.). Boca Raton, Florida: CRC Press. ISBN 978-1-4200-9084-0.
- Van Langevelde, A.; Peschar, R.; Schenk, H. (2001). “Structure of β-trimyristin and β-tristearin from high-resolution X-ray powder diffraction data”. Acta Crystallographica Section B Structural Science 57 (3): 372. doi:10.1107/S0108768100019121. edit
- Sharma, Someshower Dutt; Kitano, Hiroaki; Sagara, Kazunobu (2004). “Phase Change Materials for Low Temperature Solar Thermal Applications” (PDF). http://www.eng.mie-u.ac.jp. Mie University. Retrieved 2014-06-19.
- Charbonnet, G. H.; Singleton, W. S. (1947). “Thermal properties of fats and oils”. Journal of the American Oil Chemists Society 24 (5): 140. doi:10.1007/BF02643296. edit
- Trimyristin in Linstrom, P.J.; Mallard, W.G. (eds.) NIST Chemistry WebBook, NIST Standard Reference Database Number 69. National Institute of Standards and Technology, Gaithersburg MD. http://webbook.nist.gov (retrieved 2014-06-19)
- .
- . Fisher Scientific. Retrieved 2014-06-19.
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

…………

 ppm, CDCl3) 0.30 (m, 1H), 0.54-0.95 (m, 10H), 1.05-1.2 (m, 1H ), 1.39 (s, 9H), 1.89 (d, 3H J 6.8Hz), 2.86 (bs, 1H), 3.28-3.60 (m 2H ), 5.79 (s,1H), 6.24 (d,1H J 15Hz), 6.87 (s 1H), 7.0-7.15 (m 1H), 7.56-7.76 (m 4H), 7.88 (d 1H J 7.85 Hz), 7.92-8.04 (m 2H), 8.68 (d 1H J 8.25 Hz), 8.73 (d 1H J 8.25Hz); 13C NMR (
ppm, CDCl3) 0.30 (m, 1H), 0.54-0.95 (m, 10H), 1.05-1.2 (m, 1H ), 1.39 (s, 9H), 1.89 (d, 3H J 6.8Hz), 2.86 (bs, 1H), 3.28-3.60 (m 2H ), 5.79 (s,1H), 6.24 (d,1H J 15Hz), 6.87 (s 1H), 7.0-7.15 (m 1H), 7.56-7.76 (m 4H), 7.88 (d 1H J 7.85 Hz), 7.92-8.04 (m 2H), 8.68 (d 1H J 8.25 Hz), 8.73 (d 1H J 8.25Hz); 13C NMR (
















 .
.












































 airport
airport
















 .
.









 yu rao
yu rao









 .
.
 .
.





 .
.













 .
.

 .
.
 ISKCON
ISKCON



.jpg)

















































{kind=link}
{kind=link}
{kind=link}