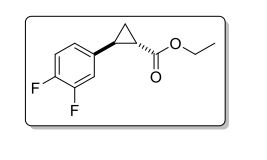
(±)-trans-ethyl 2-(3,4-difluorophenyl)Cyclopropanecarboxylate
C12H12F2O2
GC-MS (EI) m/z: [M]+ calc. for C12H12F2O2 + : 226.08; found: 226.08.
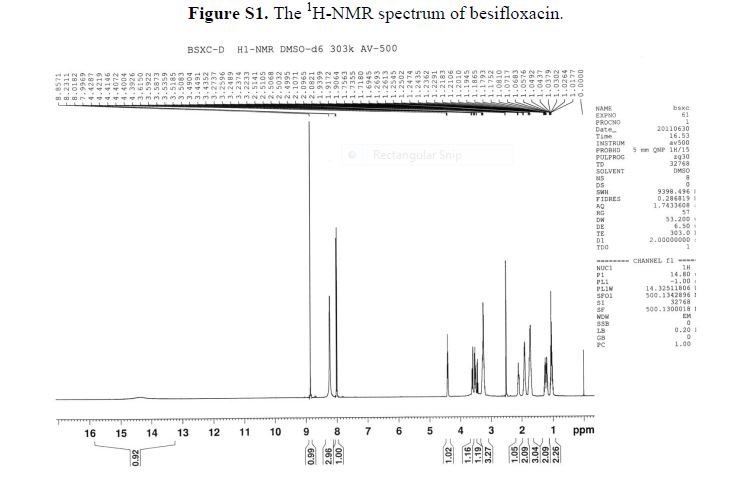
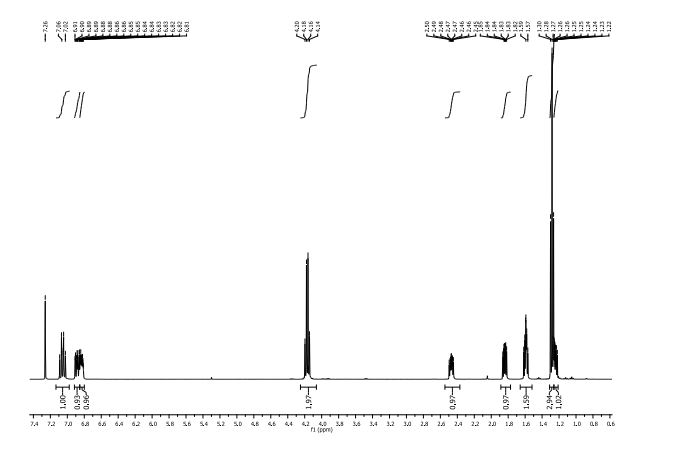
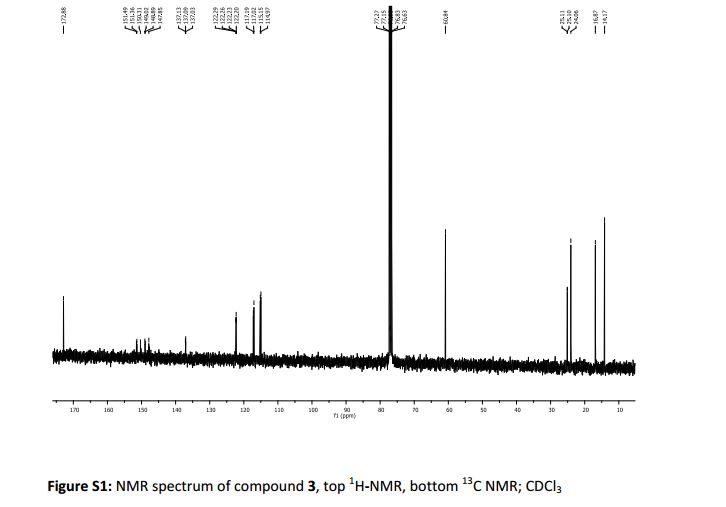
δH (400 MHz, CDCl3): 1.25 (1H, ddd, 3 J 8.4 Hz, 3 J 6.4 Hz, 2 J 4.5 Hz , 3-H); 1.28 (3H, t 3 J 6.4 Hz CH3Ethyl) 1.57-1.62 (2H, m, 3 J 9.2 Hz, 3 J 5.2 Hz, 2 J 4.5 Hz, 3-H + H2O), 1.84 (1H, ddd, 3 J 8.5 Hz, 3 J 5.3 Hz, 3 J 4.3 Hz , 2-H), 2.47 (1H, ddd, 3 J 9.5 Hz, 3 J 6.4 Hz, 3 J 4.2 Hz , 1-H), 4.17 (2H, q, 3 J 6.3 Hz, CH2Ethyl) 6.81-6.87 (1H, m, 3 J 8.5 Hz, 4 J 7.6 Hz, 4 J 2.4 Hz, 6-H’ ), 6.88 (1H, ddd, 3 J 11.5 Hz, 4 J 7.6 Hz, 4 J 2.2 Hz, 2-H’) 7.06 (1H, dt, 3 J 10.3 Hz, 3 J 8.2 Hz. 5-H’).
δc (400 MHz, CDCl3): 14.27 (CH3Ethyl), 16.84 (3-C) 24.04 (1-C), 25.14 (d, 4 J 1.4, 2-C), 60.71 (CH2Ethyl), 114.74 (d, 2 J 19 Hz, 2-C’), 117.09 (d, 2 J 18 Hz, 5-C’), 122.25 (dd, 3 J 6.1 Hz, 4 J 3.4 Hz, 6- C’), 137.06 (dd, 3 J 6.1 Hz, 4 J 3.4 Hz, 1- C’), 149.2 (dd, 1 J 248 Hz, 2 J 13 Hz, 4-C’) 151.32 (dd, 1 J 249 Hz, 2 J 12.5 Hz, 3-C’) 172.87 (Ccarbonyl).
[ ] 20 a D = -381.9 (c 1.0 in EtOH) for (1R,2R)-3, ee = 95%

In this study a batch reactor process is compared to a flow chemistry approach for lipase-catalyzed resolution of the cyclopropanecarboxylate ester (±)-3. (1R,2R)-3 is a precursor of the amine (1R,2S)-2 which is a key building block of the API ticagrelor. For both flow and batch operation, the biocatalyst could be recycled several times, whereas in the case of the flow process the reaction time was significantly reduced.
Comparison of a Batch and Flow Approach for the Lipase-Catalyzed Resolution of a Cyclopropanecarboxylate Ester, A Key Building Block for the Synthesis of Ticagrelor

“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
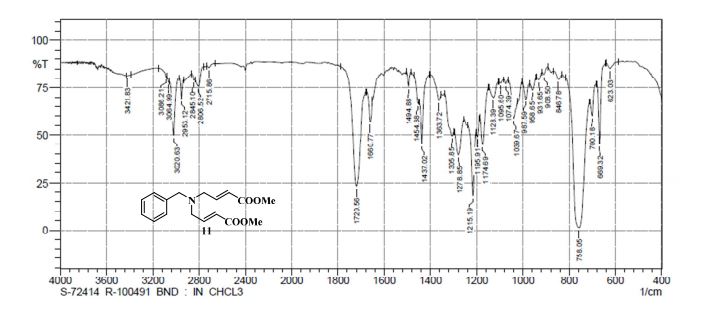
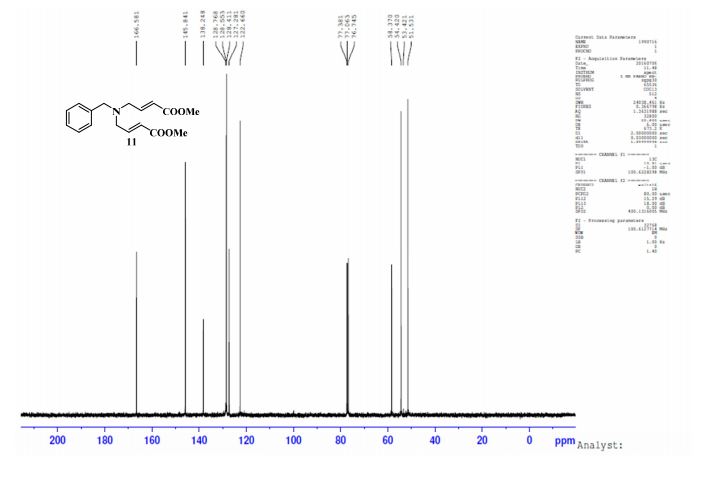
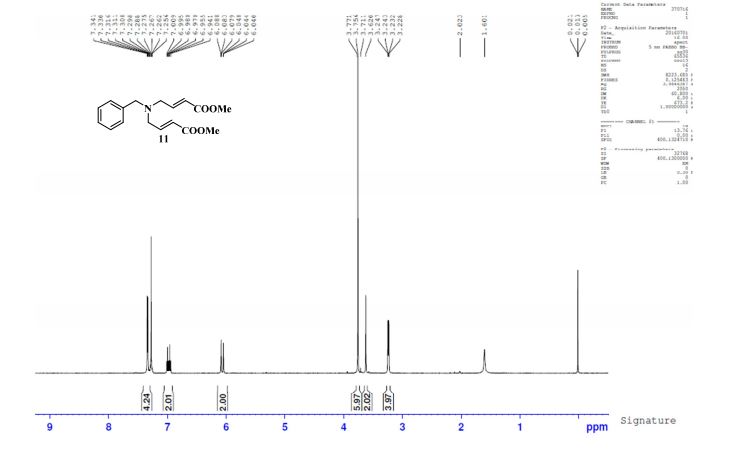
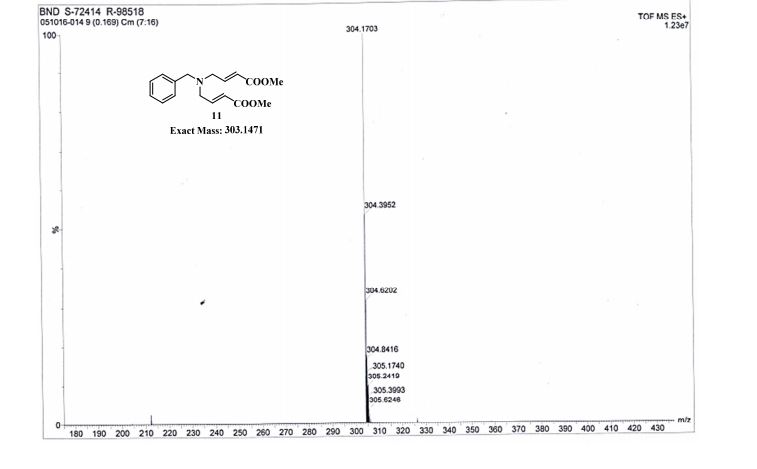

 gene.com
gene.com



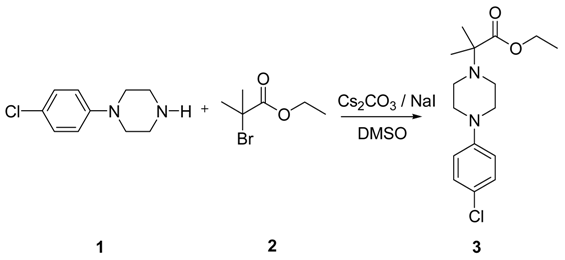


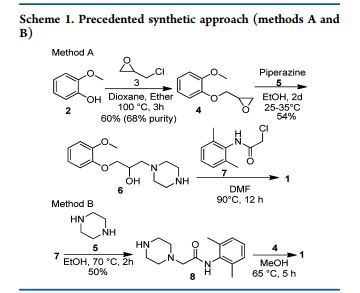











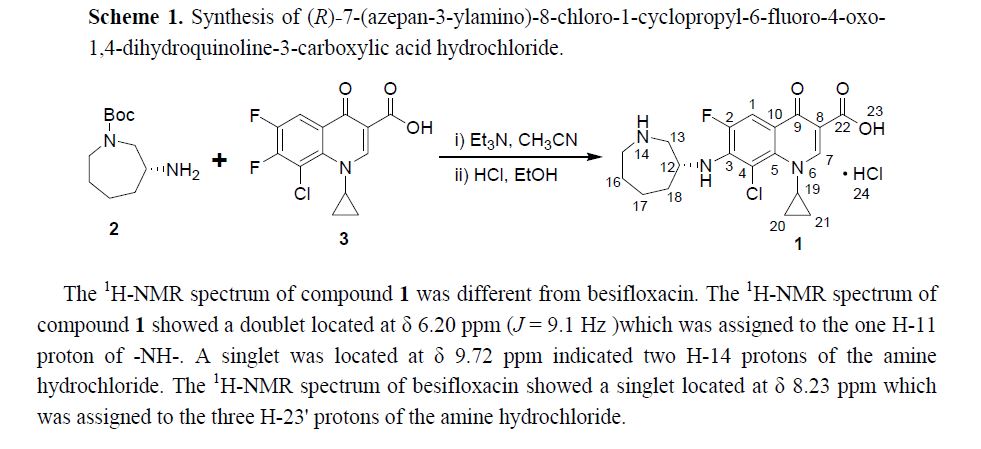
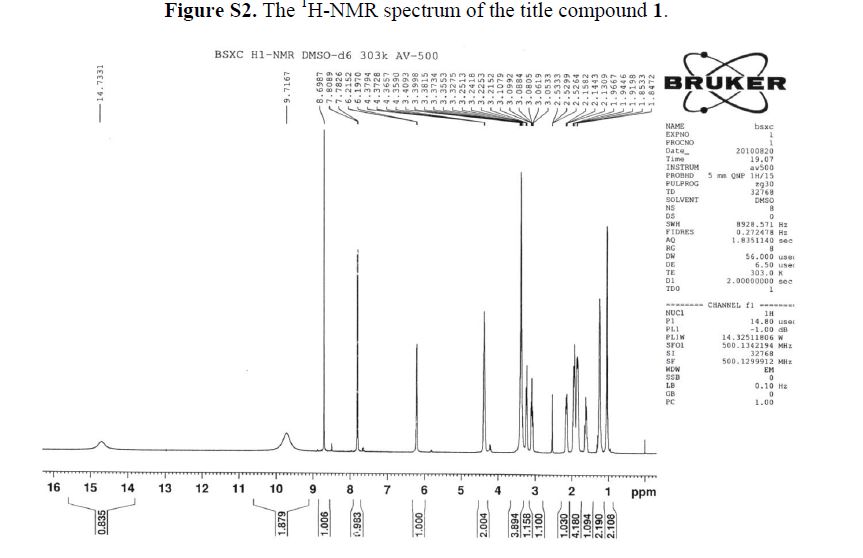
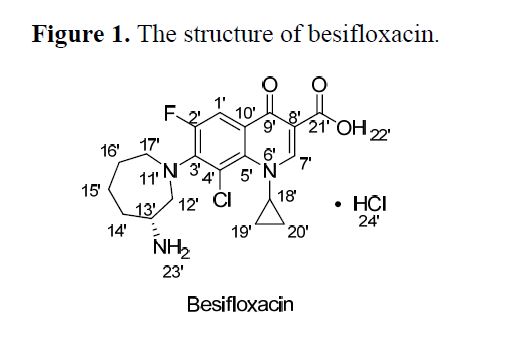
 BESIFLOXACIN
BESIFLOXACIN