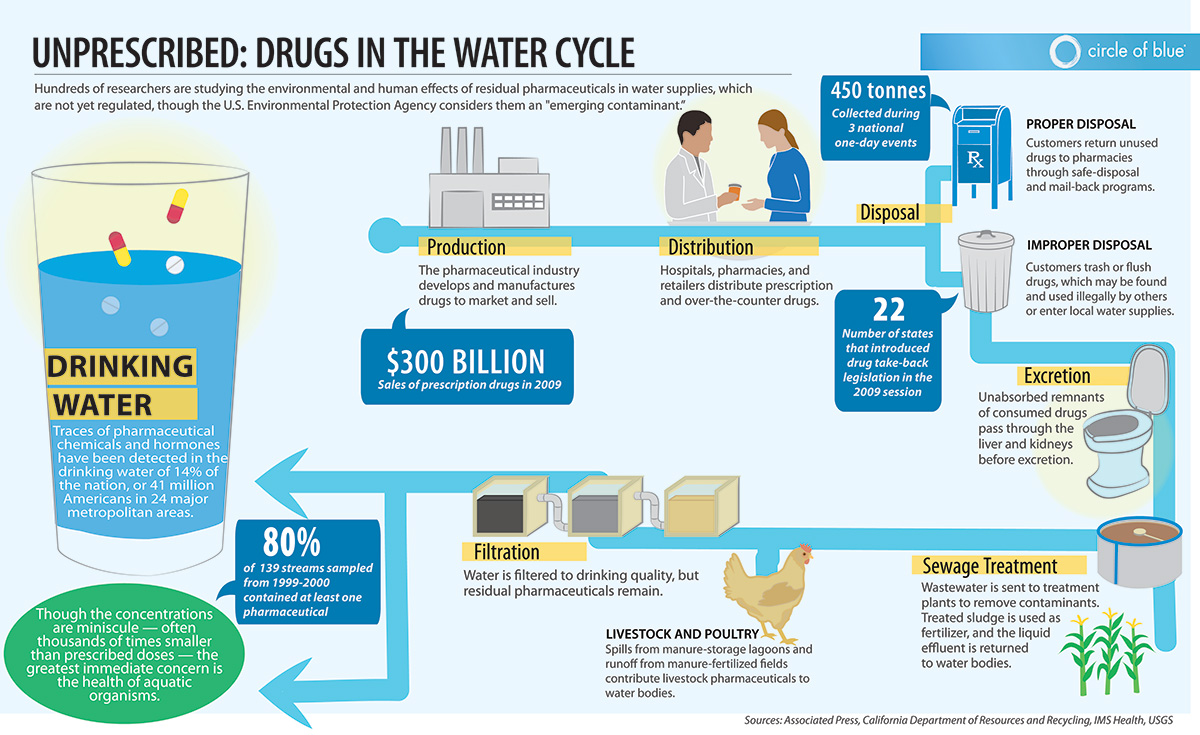
Generics: The US Food and Drug Administration (FDA) recently published a new Guidance regarding Prior Approval Supplements (PAS). Read more about FDA´s Guidance for Industry “ANDA Submissions – Prior Approval Supplements Under GDUFA“.
http://www.gmp-compliance.org/enews_05634_Generics-FDA%B4s-New-Guidance-on-Prior-Approval-Supplements_15721,Z-RAM_n.html
On October 14, 2016, the US Food and Drug Administration (FDA) published a new Guidance regarding Prior Approval Supplements (PAS).
FDA says that “this guidance is intended to assist applicants preparing to submit to FDA prior approval supplements (PASs) and amendments to PASs for abbreviated new drug applications (ANDAs)”.
Specifically, the guidance describes how the Generic Drug User Fee Amendments of 2012 (GDUFA) performance metric goals apply to:
- A PAS subject to the refuse-to-receive (RTR) standards;
- A PAS that requires an inspection;
- A PAS for which an inspection is not required;
- An amendment to a PAS;
- Other PAS-related matters.
GDUFA is designed to speed the delivery of safe and effective generic drugs to the public and reduce costs to industry. That requires that FDA and human generic drug manufacturers meet certain requirements and commitments. “FDA committed to review and act on a certain percentage of PASs within a specified period from the date of submission for receipts in fiscal year (FY) 2015 through FY 2017. The percentage of PASs that FDA has committed to review and act on increases with each fiscal year; the deadlines for review also depend on whether consideration of a PAS requires an inspection.”
Changes to an approved application:
The criteria laid down in FDA regulations for submitting information as a PAS (major change), as a Changes Being Effected-Supplement (CBE-supplement, moderate change), or in an annual report (minor change) were not changed by GDUFA.
Timelines depending on inspections for PAS submissions:
The GDUFA goal date for a PAS depends on whether the PAS requires an inspection. If a PAS does not require an inspection, the goal date is 6 months from the date of submission; but if a PAS requires an inspection, the goal date is 10 months from the date of submission. An initial goal date of 6 months occasionally may change to a 10-month goal date if, during the review, FDA determines an inspection is necessary. If an amendment is made to a PAS, the GDUFA goal date associated with that PAS may be revised. FDA strongly recommends that, at the time of submission, a supplement should be complete and ready for a comprehensive review.
Submission of Supplements:
The following information should be provided on the first page of the PAS:
- A statement indicating whether the PAS is for a new-strength product;
- A statement indicating whether the submission is an amendment to a PAS, and if so the corresponding tier classification;
- A statement indicating whether the PAS contains any manufacturing or facilities changes;
- A list of the specific review disciplines to review the PAS (Chemistry, Labeling, DMF, Bioequivalence, Microbiology, or Clinical);
- If expedited review is requested, the label Expedited Review Request should be placed prominently at the top of the submission. The submission should include a basis for the expedited review request.
It is possible to submit multiple PASs for the same chenge as “grouped supplements”. These are submitted to ANDAs by a single applicant for the same chemistry, manufacturing, and controls (CMC) change to each application. Because the grouped supplements are being reviewed together, generally they will have the same GDUFA goal date. Although the submissions are considered a group, each supplement in the group is considered its own individual submission and therefore would require a GDUFA PAS fee for each ANDA identified in the group.
Alternative Submissions:
- Identify a lead ANDA for a group of PASs (only one fee is paid, or fewer than all the fees for the group are paid);
- For some changes (e.g., widening of an approved specification or introduction of a new API supplier) once a PAS is submitted and approved, subsequent supplements for the same change to other ANDAs may be classified as CBE-30s;
- A comparability protocol submitted in a PAS to an ANDA for a specific drug product, once approved, may justify a reduced reporting category for the same change in subsequent supplements to that ANDA.
If FDA finds that a supplement submitted as a CBE supplement should have been submitted as a PAS, it will notify the applicant. The applicant is not required to withdraw the CBE supplement because when FDA sends a letter explaining that the applicant’s submission is not accepted as a CBE supplement, FDA administratively closes the CBE supplement, and it is considered withdrawn. The applicant may resubmit the supplement as a PAS for FDA approval before distribution of the drug product, along with the required GDUFA user fee. The GDUFA performance metric goals and applicable user fees will apply to that PAS and the GDUFA review clock will start from the date of submission of that PAS.
For more information please see the FDA Guidance for industry “ANDA Submissions – Prior Approval Supplements Under GDUFA“.
///////////Generics, FDA, New Guidance, Prior Approval Supplements