Pentacyclo[4.2.0.02,5.03,8.04,7]octane
CAS 277-10-1
Cubane (C8H8) is a synthetic hydrocarbon molecule that consists of eight carbon atoms arranged at the corners of a cube, with one hydrogen atom attached to each carbon atom. A solid crystalline substance, cubane is one of the Platonic hydrocarbons. It was first synthesized in 1964 by Philip Eaton, a professor of chemistry at the University of Chicago.[2] Before Eaton and Cole’s work, researchers believed that cubic carbon-based molecules could not exist, because the unusually sharp 90-degree bonding angle of the carbon atoms was expected to be too highly strained, and hence unstable. Once formed, cubane is quite kinetically stable, due to a lack of readily available decomposition paths.
The other Platonic hydrocarbons are dodecahedrane and tetrahedrane.
Cubane and its derivative compounds have many important properties. The 90-degree bonding angle of the carbon atoms in cubane means that the bonds are highly strained. Therefore, cubane compounds are highly reactive, which in principle may make them useful as high-density, high-energyfuels and explosives (for example, octanitrocubane and heptanitrocubane).
Cubane also has the highest density of any hydrocarbon, further contributing to its ability to store large amounts of energy, which would reduce the size and weight of fuel tanks in aircraft and especially rocket boosters. Researchers are looking into using cubane and similar cubic molecules inmedicine and nanotechnology.

Synthesis
The original 1964 cubane organic synthesis is a classic and starts from 2-cyclopentenone (compound 1.1 in scheme 1):[2][3]

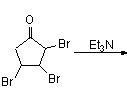
Reaction with N-bromosuccinimide in carbon tetrachloride places an allylic bromine atom in 1.2 and further bromination with bromine in pentane –methylene chloride gives the tribromide 1.3. Two equivalents of hydrogen bromide are eliminated from this compound with diethylamine in diethyl ether to bromocyclopentadienone 1.4
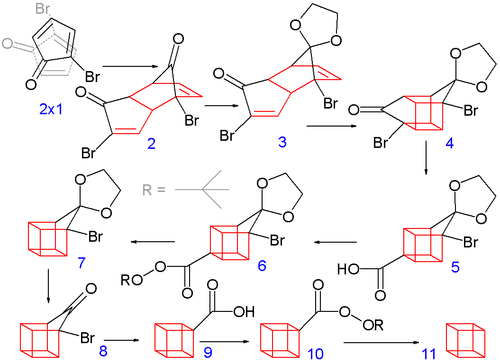
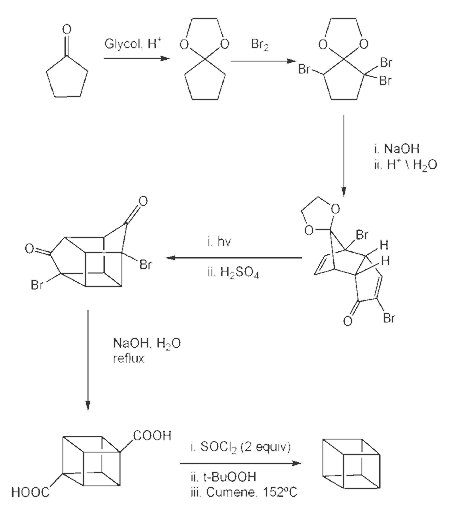
In the second part (scheme 2), the spontaneous Diels-Alder dimerization of 2.1 to 2.2 is analogous to the dimerization of cyclopentadiene to dicyclopentadiene. For the next steps to succeed, only the endo isomer should form; this happens because the bromine atoms, on their approach, take up positions as far away from each other, and from the carbonyl group, as possible. In this way the like-dipole interactions are minimized in the transition state for this reaction step. Both carbonyl groups are protected as acetals with ethylene glycol and p-toluenesulfonic acid inbenzene; one acetal is then selectively deprotected with aqueous hydrochloric acid to 2.3
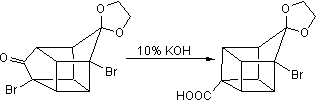
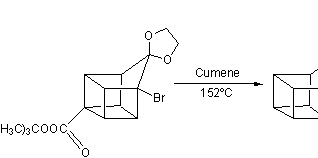
In the next step, the endo isomer 2.3 (with both alkene groups in close proximity) forms the cage-like isomer 2.4 in a photochemical [2+2] cycloaddition. The bromoketone group is converted to ring-contracted carboxylic acid 2.5 in a Favorskii rearrangement with potassium hydroxide. Next, the thermal decarboxylation takes place through the acid chloride (with thionyl chloride) and thetert-butyl perester 2.6 (with t-butyl hydroperoxide and pyridine) to 2.7; afterward, the acetal is once more removed in 2.8. A second Favorskii rearrangement gives 2.9, and finally another decarboxylation gives 2.10 and 2.11.
The cube motif occurs outside of the area of organic chemistry. Prevalent non-organic cubes are the [Fe4-S4] clusters found pervasively iron-sulfur proteins. Such species contain sulfur and Fe at alternating corners. Alternatively such inorganic cube clusters can often be viewed as interpenetrated S4 and Fe4 tetrahedra. Many organometallic compounds adopt cube structures, examples being (CpFe)4(CO)4, (Cp*Ru)4Cl4, (Ph3PAg)4I4, and (CH3Li)4.



It was mentioned previously that cubane was first prepared in 1964 by Dr. Philip E. Eaton. He was partnered by Thomas W. Cole and together they successfully completed the first synthesis, shown schematically below:
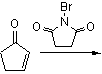
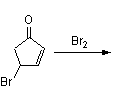
![Reactive enough to undergo dimerisation via a [4+2] cycloaddition reaction to give the ENDO cycloadduct](http://www.chm.bris.ac.uk/webprojects2004/hook/synthesis/5.gif)
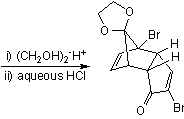
![Photochemical energy is required to promote the [2+2] intramolecular cycloaddition reaction.](http://www.chm.bris.ac.uk/webprojects2004/hook/synthesis/7.gif)
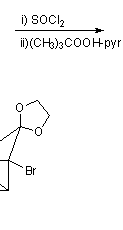
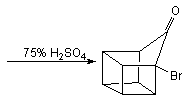
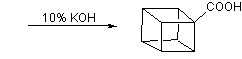
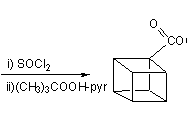
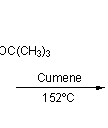
This, however, was soon simplified by N.B.Chapman who condensed the process to give cubane-1,4-dicarboxylic acid in five steps and so cubane in six:
n 1966 J C Barborak et al discovered yet another new synthesis of cubane. It was slightly unconventional in the fact that it utilised cyclobutadiene as a key substance to the process. Before this,cyclobutadiene was usually unavailable for the purposes of organic chemistry due to it’s instability. The shorter synthesis is shown below:
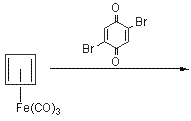
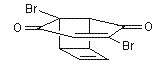
![Irradiation, in benzene, with a mercury lamp initiates the intramolecular [2+2] cycloaddition reaction.](http://www.chm.bris.ac.uk/webprojects2004/hook/synthesis/barboraksynthesis_r4_c7.gif)
Since the synthesis of the cubane-1,4-dicarboxylic acid has become shorter and easier, a new decarboxylation method has also devised to give increased yields of the final cubane product. This has allowed the scale of production reach multikilogram batches in places (Fluorochem in California and EniChem Synthesis in Milan) eventhough cubane and its derivatives remain expensive to purchase.
Cuneane may be produced from cubane by a metal-ion-catalyzed σ-bond rearrangement.[4][5]
Cubane is a unique molecule for its extraordinary C8 cage, very high symmetry,exceptional strain and unusual kinetic stability. The particular appeal of cubane,referred to as a landmark in the world of impossible compounds, stems from therehybridization of the carbon atoms away from the canonical sp3 configuration,that is required to bound together eight CH units in a cubic framework.There is now a revival of interest on the chemistry of cubane and its functionalized derivatives,triggered by potential applications as high-energy fuels, explosives and propellantsand intermediates in pharmaceuticalpreparations.Let us now discover the synthesis and properties of this landmark molecule of impossible chemistry
Cubanehas the highest strain energy (166kcal/mol) of any organiccompounds available in multi gram amount. It is a kineticallystable compound and only decomposite above 220 Celsius Degree.It is also one of the most dense hydrocarbons ever know.However, although many physical properties of cubane have been measured, in1980 and before, cubane was considered just a laboratory curiosity of interest only to academics.It changed, in early 1980s when Gilbert of U.S ArmyArmament and Development Command (now ARDEC) pointed out that cubane’svery high heat of formation and its exceptionally high density could make certain cubanederivatives important explosives. The effectiveness of an explosive is dependent on the energentics of the decomposition reaction,the number of moles and molecular weight of the gaseous products and also the density. The effectiveness of an explosive is dependent on the energentics of the decomposition reaction,the number of moles and molecular weight of the gaseous products and also the density.
The more mols of of an explosive that can be packed into the limited volume the better. .
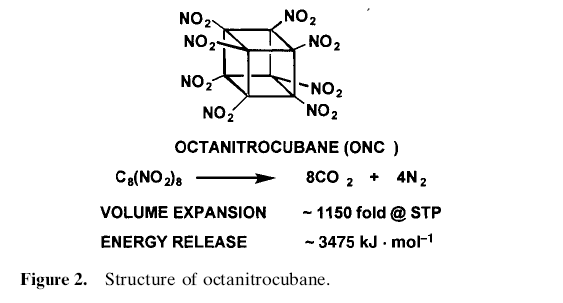
Highly nitrated cubanes can be predicted to be very dense and very powerful explosives.
Octanitrocubane is calculated to be 15~30%more powerful than HMX.
| Cubane, which CA index name is Pentacyclo[4.2.0.02,5.03,8.04,7]octane (7CI,8CI,9CI),has exceptional structure, strain and symmetry and it is a benchmark in organic chemistry.It has been studied extensively and much of its properties has been published.Some of the physical properties are given at right hand table. |
|
.png&container=blogger&gadget=a&rewriteMime=image%2F*)
The C-C bond length is a bit longer than obtained in the original X-ray structure determination by
Fleischer in 1964. There is not much difference between this bond length and the
C-C bond length in a simple cyclobutane.

SYNTHESIS
The cubane system was first synthesized over 35 years ago by Philip Eaton and Tom Cole.
It is a highly symmetric cubic cage structure having carbon atoms at the vertices of a cube.
The synthesis needs to go through brombromocyclopentadienone
dimer I and cubane-1,4,dicarboxylic acid. It is a marvel scheme of economy and simplicity.
With only minor modification, this procedure remains to this day the best available
method for large-scale synthesis of cubane-1,4,dicarboxylic acid.



The stereospecific in situ [4 + 2] (Diels-Alder) cyclodimerization of 4-bromocyclo-pentadienone
is the key in this kinetically controlled synthesis. However, it is still a tricky matter
and a few years later after this synthesis is published, N.B.Chapman et al in England following up
this work and improved this synthesis.
Why cubane is stable?
The reason for this, unappreciated at the time of the early predictions of instability,
is that there are no kinetically viable paths along which cubane can rearrange thermally.
On one hand, orbital symmetry considerations raise the energy of concerted two-bond ring
opening reactions. On the
other, there is little to be gained by breaking just one bond as there is concomitantly
only a small change in geometry, and the resulting biradical is still very strained.
Functional group transformation
| Functional groups on the cubane system generally behaves very well.Functional group transformation can be applied successfully.For example, the preparation of 1,4-dinitrocubane from cubane-1,4-dicarboxylic acid.(The mechanism is provided on the right hand side.) Classical methodology is used here. |

Substitution on the cubane framework is fairly easy done by the cubyl radical.
However, the problem is such that a mixture of products are obtained.
Thus, to achieve controlled substitution on the cubane framework,
we need to carefully study the chemistry of the cubane system.
The improvement in synthesis of
cubane-1,4-dicarboxylic acid

This is the improved synthesis by N.B Chapman et al in England.
Basically the improvement is such that the
2-bromocyclopentadienone could be made easily and undergoes spontaneous dimerization.
The rest of the reaction is the same as the original one.
This synthesis now is scaled up and is conducted in small pilot plants by
Flurochem in California and EniChem Synthesis in Milan.
This method is much more superior than the old method. It is introduced by
Derek Barton et al and use the radical-induced decomposition of diester which can be
prepared easily from cubane-1,4-dicarboxylic acid.
This method is much more superior than the old method. It is introduced by
Derek Barton et al and use the radical-induced decomposition of diester which can be
prepared easily from cubane-1,4-dicarboxylic acid
Cubane is a colorless solid. It melts at 130- 131°C, and decomposes above the melting point.
It is soluble in CS2, CC14, CHC13, and benzene.
Spectra were obtained from 400 to 3600 cm-l with a Beckman IR-12 spectrophotometer.
The lower limit was set by KBr cell windows. In addition a thick deposit of do was
measured down to 200 cm-lin a Csl cell. Since no infrared bands were found, the range
200-400 cm-l was not examined for the other compounds.
The spectral slit widths were 1-2 cm-l in all cases.
In the infrared spectrum, there are only noticeable absorptions in the region from
4000 to 660 cm-1appear at 300,1231, and 851 cm-1.


Generally, for single-line proton magnetic resonance spectrum, the one
and only absorption appears at chemical shift=6.0ppm.
Originally there was doubt whether cubane does exist.
The geometry at each carbon atom is far from tetrahedral.
Only later, we found out that there is no kinetically viable paths exist for
the thermal rearrangement of cubane.
At same time, orbital symmetry considerations shows that
the energy of concerted two-bond ring-opening reactions is very high.
There will be very little gain in energy by breaking just one bond, as the
concomitant change in geometry is small, and the resulting biradical is still very strained
In 1964 Fleischer showed that cubane forms a stable solid at room temperature with a
crystalline structure composed of cubane molecules occupying corners of the rhombohedral
primitive unit cell (space group R3). The cubic molecular geometry gives the solid many unusual
electronic,structural, and dynamical properties compared to the other hydrocarbons.
For example, solid cubane has a relatively high melting point temperature about 405 K! and a
very high frequency for the lowest-lying intramolecular vibrational
mode (617 cm-1). Recent work related to cubane has focused on solid cubane and cubane based
derivatives.Because of relatively weak intermolecular interaction the cohesive energy relative
to the constituent C8H8 is expected to be small, and most of the physical properties of
solid cubane are dominated by the properties of the C8H8molecule.

Pharmaceutical aspect of cubane
Because the cubane frame is rigid, substituent have precise spatial relationships to each another.
The distance across the cubane (the body diagonal) is almost the same as that between the para
positions of the benzene ring. On cubane, on can add substituents in the “benzene plane”, as
well as above and below it, so to speak. This offers fascinating position possibilities for
the synthesis of new pharmaceuticals. A number of cubane derivatives have already
been obtained which shows interesting activity in anti-AIDS and anti-tumor screens.
Although the activity or the toxicity balance of cubane is yet not know, the cubane
system is not inherently toxic. Most of cubanes are biologically innocuous.
The research of cubane pharmaceutical has just began. At least now,
cubane is a biologically stable, lipophilic platform on which the chemist
can install a wide range of substituents in a variety of well defined special relationships.
Developments in drug design programs should allow the judicious choice.

Dipivaloylcubane: a cubane derivatized with keto, cyano, and amide groups,
shown on the left- exhibits moderate activity against human immunodeficiency virus (HIV),
which causes AIDS, without impairing healthy cells.
Polymers of cubane:
Optically transparent cubanes and cubylcubanes have been proposed as building
blocks for rigid liquid-crystal compounds. UV active cubanes, for example cubyl ketones,
are readily transformed photochemically into coloured cyclooctatetraenes;this transformation
can be used to permanent information storage.
Another example of UV active cubane, which can be used to synthesis liquid crystals.

Polymers with cubane in the backbone or as a pendant group along a polymer chain is
focused now.
The cubane subunits in these polymers can be rearranged easily to cycloctatetraenes.
It is expected that polycyclooctatetra can be converted in to polyacetylenes by
the way of ring-opening metathesis polymerization. The polyacetylenes will have properties
which are enhanced by the chain being intrinsically part of another polymer.
These properties including stability and extrudability and etc. A example is shown below:

Cubane derivative could be the structural basis for a class of intrinsic small gap polymers.The small gap polymer could present intrinsic good conductivity without doping,good nonlinear optical and photoelectric properties.Investigation of oligamers with up to six units of a conjugated unsaturated cubane derivative,where all the hydrogen were removed, is carried out.The table below shows that the gap values in eV by EHT and PM3.These values suggest to us that these structures could be used to design a newclass of polymers with very small gap. |
Explosive and fuels:
In the early 1980s Everett Gilbert of the U.S. Army Armament Research and Development
Command (now ARDEC) pointed out that the nitrocarbon octanitrocubane (ONC),
then unknown, has a perfect oxygen balance, and in light of the properties of the
parent hydrocarbon cubane should have a very high heat of formation per CNO2 unit
and an exceptionally high density as well. His colleagues Jack Alster, Oscar Sandus
and Norman Slagg at ARDEC provided theoretical support for Gilbert’s
brilliant insight and estimated that ONC would have a detonation pressure
significantly greater than HMX. Later, both statistical and computational
approaches predicted a density of 2.1 ± 2.2 g /cm3 for octanitrocubane,
greater than any other C, N, O compound.

Is Cubane a really good explosives?
Quantitative evaluation of the potential of a candidate explosive before synthesis is very difficult.
Currently, estimation of energetic properties relies on the empirically derived Kamlet and Jacobs
equations:

In these equations the heat released by the decomposition, the number of moles of gas produced,
and the molecular
weight of these gases are all critical factors. Density too is crucial.
Obviously, the more molecules of a high-energy material that can be packed into the limited
volume of a shell or rocket the better. Less obvious, but more important, density affects the
detonation velocity of an explosive.
This is a specialized “linear” rate of reaction that ranges from 5 to 10 km/s in
explosives and affects the maximum detonation pressure, a direct measure of the
power of an explosive. For a given explosive, the detonation pressure is proportional
to the square of its density, so great effort is made to obtain the highest density form
of any particular explosive.
Quantitative evaluation of the potential of a candidate explosive before synthesis is very difficult.
Currently, estimation of energetic properties relies on the empirically derived Kamlet and Jacobs
equations:
In these equations the heat released by the decomposition, the number of moles of gas produced,
and the molecular
weight of these gases are all critical factors. Density too is crucial.
Obviously, the more molecules of a high-energy material that can be packed into the limited
volume of a shell or rocket the better. Less obvious, but more important, density affects the
detonation velocity of an explosive.
This is a specialized “linear” rate of reaction that ranges from 5 to 10 km/s in
explosives and affects the maximum detonation pressure, a direct measure of the
power of an explosive. For a given explosive, the detonation pressure is proportional
to the square of its density, so great effort is made to obtain the highest density form
of any particular explosive.
Numerous nitro compounds are employed commonly as military and commercial explosives.
There is a continuing search for more powerful and less shock-sensitive examples.
Such materials are also sought as potentially useful fuels and propellants.
Most interest is focused on high-density organic compounds that contain all of the
elements needed for combustion to gaseous products in the absence of air.
Nitrocubanes carrying five or more nitro groups contain enough oxygen to oxidize
all constituent carbon and hydrogen atoms to gaseous CO, CO2, or H2O.
Each of these, along with N2, “explodes” from the solid to 12 gaseous molecules.
The expansion from the dense solid to a lot of gas (much expanded by the released heat)
produces the desired effect in propellants and explosives. ONC has a “perfect”
oxygen balance and would produce (were the detonation completely efficient)
eight molecules of carbon dioxide and four of dinitrogen. As ONC has no
hydrogen, no water forms when it burns; when used as propellants such zero-hydrogen
compounds leave little or no visible smoke (steam) in the plume behind the rocket;
such “low-signature” rockets are difficult to track.

On application of the Kamlet and Jacobs equations led ARDEC to predict that
octanitrocubane would be a very much better explosive (Table 1) than the classic
C-nitro compound trinitrotoluene (TNT), perhaps 15±30% better than the nitramine
HMX (the most powerful, commonly used military explosive), and at least competitive
with (and perhaps less shock-sensitive than) the newest experimental explosive CL-20

SYNTHESIS:(1)
The high strain that the cubane framework is under has already been highlighted. The researchers had to very cautiously attach a nitro group to each of the corners of the cube in order to make the desired product. The insertion of the first four nitro groups could be done by manipulating functional groups:
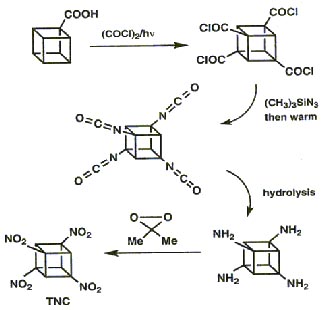
The key intermediate, cubane-1,3,5,7- tetracarboxylic acid (TNC), was obtained by clever application of the Brown-Kharasch photochlorocarbonylation to cubane mono-acid.
The addition of four further nitro groups proved far more difficult and new methodologies had to be developed, specifically the process of interfacial nitration. This method was used successfully to convert the sodium salt of TNC to pentanitrocubane (PNC) and then hexanitrocubane (HNC), both are stable materials.
Interfacial nitration, however, proved deficient for further nitration of HNC and again new experimental methodology had to be developed for its successful conversion to heptanitrocubane (HpNC):
Addition of excess NOCl to a solution of the lithium salt of HpNC in dichloromethane at -78°C gave the long-sought ONC:
DIFF TYPES

For the last planned post in my Unnatural Products series, I’m going to write about Eaton’s 1981 synthesis of pentaprismane.[A] At the time, unnatural hydrocarbons were hot targets, and as the next largest prismane on the list this target was the subject of much research by groups around the world. Perhaps Eaton’s biggest rivals were the groups of Paquette and Petit, and in fact all three had, at various times, synthesised hypostrophene as an intended precursor to the target.

Unfortunately, the ‘obvious’ [2 + 2] disconnection from pentaprismane turned out to be a dead end and the photochemical ring closure was unsuccessful. The 1970s and early 1980s saw the publication of a number of other similarly creative, but sadly ill-fated, approaches based on various ring contractions, and the compound gained a well-earned reputation for extraordinary synthetic inaccessibility.
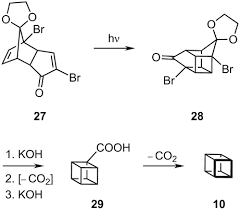
Eaton’s route began, as with the cubane and dodecahedrane syntheses previously covered in this series, with a Diels-Alder reaction. The diene used was the known tetrachlorocyclopentadienone acetal shown that upon heating neat with benzoquinone produced the endo adduct shown in excellent yield. Next, an even higher yielding photochemical [2 + 2] reaction was used to close the cage-like structure by cyclobutane formation. Treatment with lithium in liquid ammonia simultaneously reduced both ketones and removed all four chlorine atoms. The resulting diol was converted to the ditosylate, which, under carefully controlled conditions with sodium iodide in HMPA, underwent a mono-Finkelstein reaction to give the iodotosylate shown. When this was treated with t-BuLi halogen-lithium exchange, followed by an extraordinary fragmentation, gave a diene reminiscent of hypostrophene shown above. However, the extra carbon atom in the skeleton made all the difference, and unlike the parent compound, this did undergo a [2 + 2] cycloaddition when exposed to UV light. Finally, acetal hydrolysis gave homopentaprismane in 34% yield from benzoquinone, putting the group a single ring contraction from victory.[B]

With significant amounts of homopentaprismanone in hand, the group now intended to employ the transformation that had been the cornerstone of their cubane synthesis – the Favorskii rearrangement. Unfortunately, this required the introduction of a leaving group in the ketone α-position, a transformation made incredibly difficult due to the strained system and Bredt’s rule, which prevented enolisation.[C] Eventually a six-step sequence (!) to introduce a tosyloxy group was devised, beginning with a Baeyer-Villiger reaction using m-CPBA. A remarkable CH oxidation with RuO4, generated in situ, then gave the hydroxylactone. Treatment of this with diazomethane gave the corresponding δ-ketoester in almost quantitative yield. The group then reformed the starting norbornane-like bridge through use of an unusual acyloin type reaction effected by treatment with sodium in liquid ammonia. Finally, oxidation of the secondary alcohol and tosylation gave the Favorskii precursor, apparently preparable in muti-gram quantities.

Treatment with aqueous potassium hydroxide solution effected Favorskii rearrangement in excellent yield, especially considering that this was the first time the elusive pentaprimane ring system had been prepared. Finally, Eaton used the three-step decarboxylation he had developed for cubane to remove the extraneous acid and give pentaprismane in 18 steps. Awesome.[D]
 References and suchlike
References and suchlike
- A J. Am. Chem. Soc., 1981, 103, 2134. Much like Eaton’s seminal cubane paper, the title is a single word, ‘Pentaprismane’. I love the lack of hype.
- B Although Petit had prepared this compound a full decade earlier, his approach relied on a cycloaddition of the difficult to prepare cyclobutadieneiron tricarbonyl with the acetal of tropone, and proved difficult to scale up. In fact, in his own paper Eaton rather directly described it as ‘conceptually fascinating [but] useless synthetically’.
- C Eaton uses the phrase ‘invasion at the bridgehead’, which I find delightfully evocative. Makes it sound like a second world war campaign. Apparently the group initially planned, in spite of Bredt’s rule, to deprotonate the bridgehead position, relying on inductive stabilisation of the anion rather than enolate formation, but were unable to do so.
- D Pentaprismane is the most recent of three prismanes synthesised to date, the other two being cubane, and triprismane. Although I think triprismane looks quite silly, it was actually synthesised some 8 years previouslyby T. J. Katz in far fewer steps. Go figure.


The Amide Activating Group
The very first step of cubane frame substitution will be the activation of the cubane frame.
This can be done by amides. The idea is derived from the similarities between cubane and arenes.
Both of them have C-H bonds with enhanced s character ( see structure),
and in both the adjacent (ortho) substituents are forced to be coplanar.
A more specific example is the cubane-N,N-diisopropyl carboxamide
reacts with excess lithium tetramethylpiperidide (LiTMP) in THF solvent.
About 3% of the deuteriation products obtained.
The diisopropyl amide activating group is used because it is inert to the amide
bases employed for ortho metalation. Although there is a problem, there is
difficulty in hydrolyzed it the corresponding carboxylic acid.
The problem is finally solved by using borane reduction followed by the oxidation
of the amine so produced with dimethyldioxirane or potassium permanganate (in large scale).
Transmetalation is the basis of a complete synthetic methodology for the preparation
of a great variety of the substituted cubanes.
In order to make the substitution productively, a way must be found to
make use of the small amount of anion in the equilibrium with the starting material.
Mercury salt is used here as an effective anion trap and very little starting material remain unreacted.
The mercury for lithium transmetalation resulted in nearly complete conversion of the
starting material by drawing the lithiation equilibrium to the right.

The amide group is important in stabilizing the intermediate lithiated cubane,
but not the mercuriated compound. Once the lithium is replaced by mercury,
the amide group is again able to assist removal of another ortho-hydrogen atom.
In the end, the complex ortho-mercurated product mixture obtained was
simplified by treatment with elemental iodine.
The iodine cleavage of the carbon-mercury bonds 2-iodo and 2,6-diiodo derivatives
of the starting amide in72% and 15% respectively
Cubyl Grignard Reagents
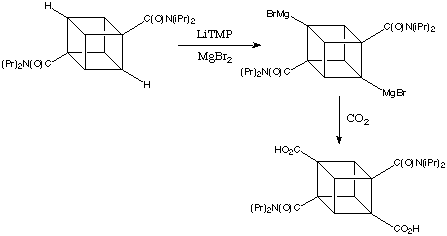
From transmetalation, a reverse transmetalation was also developed, which is basically adding Grignard reagent to the mercuriated cubane instead of the iodine. However, these processes have a great main disadvantage, the mercury is highly toxic. Thus, scale up of this method was limited.
In 1988, Bashir-Hashemi introduced transmetaltion with magnesium salts and thereby provided easy access to cubyl mono-and bis-Grignard reagents. It is a reaction of cubane diamide with an excess of LiTMP/MgBR2 in THF and quenching with I2 gave diiodocubane diamide of 72% yield.
The effect of the presence of electron withdrawing group – Cyanide
When electron withdrawing group such as cynate present, they stabilize both intermediate lithiated cubanes very well. As a result, only a small amount of LiTMP is need to achieve fairly complete deprotonation even at -78°C.
The inductive effect of the cyano group clearly enhances the reaction. However, the adding of cyano groups results in competitive lithiation and a mixture of products. However, this problem can be well trackled by adding MgBr2.The product ratio was improved to 9:1 favoring carboxyliation ortho to the amide function.
A mixture of product formed.
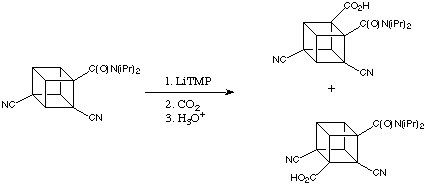
Increased selectivity by adding MgBr2
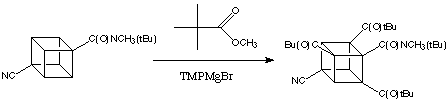
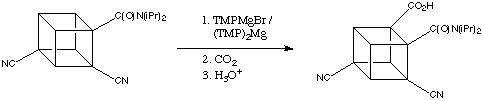
Since the reactivity of cubane metalation is enhanced greatly with presence of cyano groups, it is possible to substitute all three positions ortho to the amide in a simple reaction. For instance, 4-cyanocubanamide can be converted directly into the tri(tert-butylcarbonyl)derivative as shown below.
Through Baeyer-Villiger oxidation, ter-butyl cuybl ketones can be converted easily to the polycarboxyliated cubane.
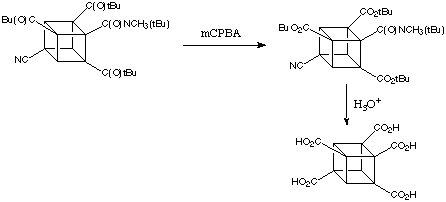
PHENYL CUBANE
From the basis idea of cubyl Grignard Reagent, phenyl cubane can be synthesised. The reaction of cubane diamide with 10 equiv of LiTMP and 4.0 equivalents of MgBr2 etherate in THF at 0°C followed by the addition of 10.0 equiv of bromobenzene, gave diphenylcubane diamide in 53% yield.
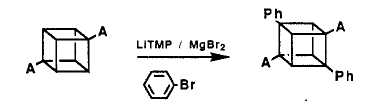
The mechanism is shown below:
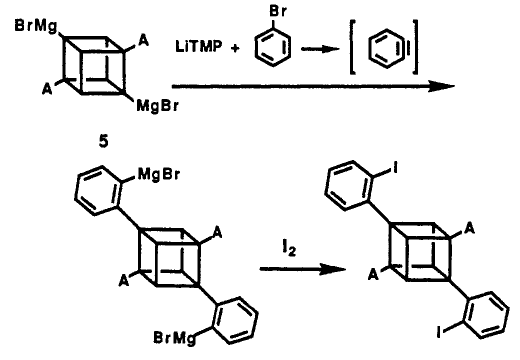
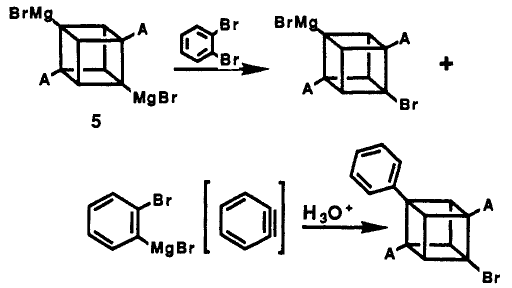
The benzyne intermediate was formed in situ from the reaction of excess of LiTMP with bromobenzene. For a similar reaction, MeMgBr is used and give 30% yield of bromo-phenylcubane diamide, the first cubane derivative containing 3 different substituents.
Now, let us look the main concern of the cubane derivatives–the nitrocubanes.
Nitrocubanes are sought to be powerful, shock-insensitive, high-density explosives. They are stable compounds with decomposition points above 200°C. Simple nitrocubane can be made from simple oxidation of amines( See Functional Group Transformation.)
If we want to add more nitro groups into the cubane nucleus, we cannot do it though transmetalation because there is unstoppable cage cleavage reactions when make adjacent nitro groups. The ab initio calculation has confirmed this destabilising effect.
We are going to discuss how to make more and more substituted nitrocubane until octanitrocubane(ONC), the ultimate power house, is synthesised.
1,3,5 trinitrocubane and 1,3,5,7 tetranitrocubane(TNC)
As we mention early, addition of nitro groups cannot be done through direct transmetalation. Thus, we need found some indirect route.
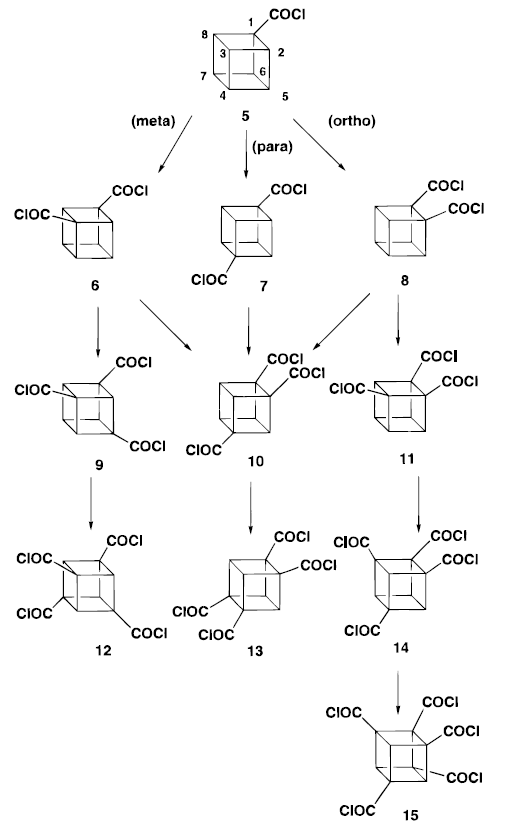
This is done by introducing a substituent on each of 3 ortho carbons and remove the ortho-activating group in the end.
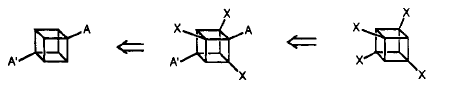
By adding a electron-withdrawing group such as a cyano group will help the case here. This choice of original substituent is important here and when cyano group is chosen, it activates the cubane nucleus without affecting the ortho directing by the diamide (for details please refer to electron-withdrawing group-cyanite).
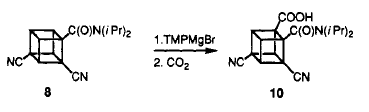
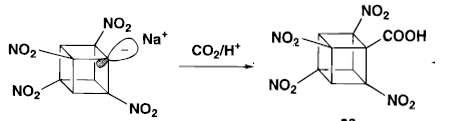
When the dicyano amide was treated with TMPMgBr in THF and quenched with CO2. The ortho (to amide) carboxylic acid was the only product.
Even when the much activated tricyanoamide is treated with TMPMgBr and CO2 ,again, the ortho position ( to amide) carboxylic acid was formed.
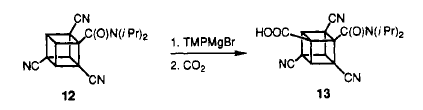
The removal of the carboxamido group is done through a smart yet tedious process. The cyano group is converted to acid group first. Then, it is reduced to alcohol by lithium aluminium hydride. At same time, the carboxamido is reduced to aminotetrol. The alcohols are protected as acetates and amino tetrol is converted to carboxylic acid. The carboxylic is then removed through Barton Decarboxylatio. A detail mechanism is provided below.
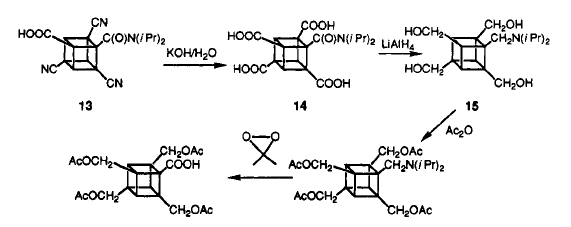
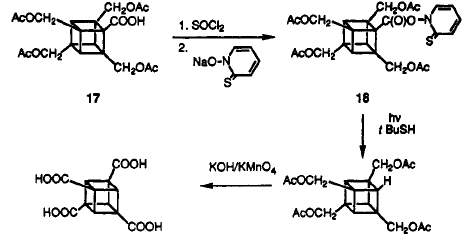
The cubane-1,3,5,7-teracarboxylic acid is converted to TNC on the mechanism as follow:
The whole process is very clever, but it is very long. Thus, in 1997, a improved synthesis method for TNC was proposed by making use of the photochemsitry.
Improved synthesis for TNC
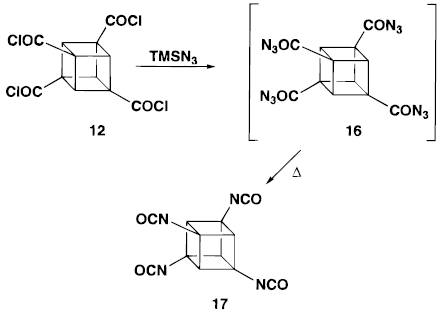
In 1993, Bashir-Hashemi showed the cubane-1,3,5,7-tetracarboxylic acid chloride can be formed by applying photochemically induced chlorocarbonyl cation( the Kharasch_Brown Reaction).
For a fast reaction, a high power Hanovia of 450 watts, medium pressure Hg was used. The favoured products are cubane tetraacid chloride shown on the right hand side. The first one, cubane-1,3,5,7-tetracarboxylic acid, made up 30% overall. This reaction conveniently prepare us the important versatile intermediate .
A detail conversion process is provided below:
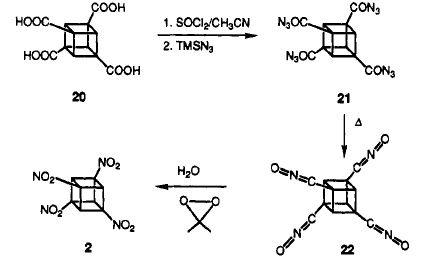
A catalyst TMSN3 is used in converting tetraacid chloride to tetracylazide. The rest is the same as the orginal reaction.
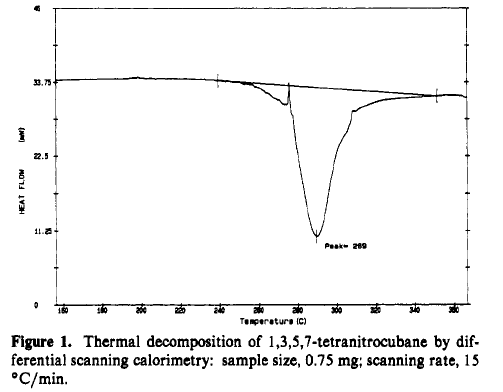
TNC is a thermodynamic powerhouse but remarkly stable kinetically. Figure 1 shows that rapid thermal decomposition doesnot start until over 250°C.
The literature was unsupportive of this optimistic view. Poor results were also obtained initially with nitrating agent such as NO2BF4, acetyl nitrate, amyl nitrate etc.
Tetranitrocubylsodium can be formed directly on treatment of TNC with sodium bis(trimethylsilyl) amide in THF at -75°C. It can react with electrophiles to provide a useful and convenient way to achieve further functionalization of cubane nucleus.
More substituted nitrocubanes-
Pentanitrocubane(PNC) and Hexanitrocubane(HNC)
PNC
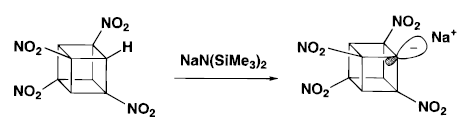
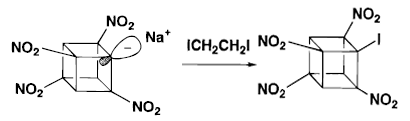
Base on the property of tetranitrocubylsodium, nitryl chloride(NO2Cl) was used to further nitrate the cubane nucleus. Treatment of NO2Cl with tetranitrocubylsodium in THF at -75°C works out 10-15% yield of pentanitrocubane(PNC). The yield increased to 30% when the solution was frozen to-180°C and allowed to warm slowly. This is called the interfacial nitration process. It is suggested that NO2Cl oxidized tetranitrocubylsodium to a radical, which made the whole reaction worked.
Base on the property of NO2Cl , N2O4 should be a better choice. The results showed that it is actually a better with 60:40 PNC to TNC ratio. The reaction is extremely clean.
PNC is colourless and highly crystalline. It is the first nitrated cubane to contain adjacent nitro groups. It behaves just TNC and other nitrocubanes, remarkly stable kinetically.
HNC
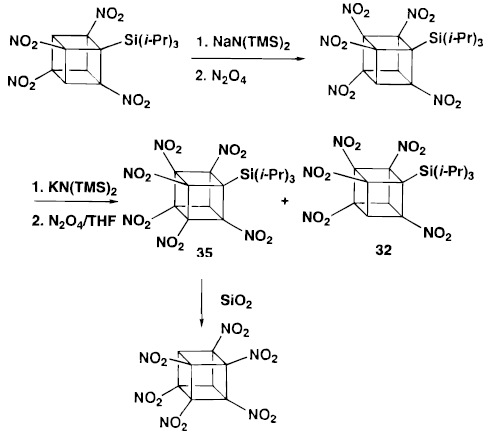
Although HNC can be prepared the same way as PNC, but the separation between PNC and HNC is extremely difficult.
However, if TIPS-substituted PNC( by N2O4 nitration from TIPS-sub TNC) react with potassium base (K(TMSN)2and the nitration with N2O4 gave a mixture of (triisopropyl) HNC and PNC in 60:40 ratio. This step is important and crucial. The separation is now possible by column chromatography on silica gel. 30% isolated yield of PURE HNC could be obtained when further treated with SiO2.
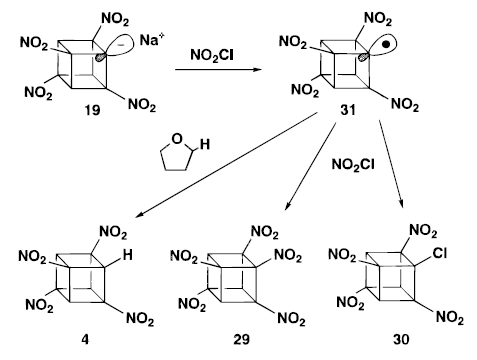
Synthesis for the last two nitro cubanes- heptanitrocubane and octanitrocubane
Interfacial nitration is not sufficient to further nitration for heptanitrocubane. Al though it is very good in deed, we need to find something which can successfully convert heptanitrocubane (HpNC).
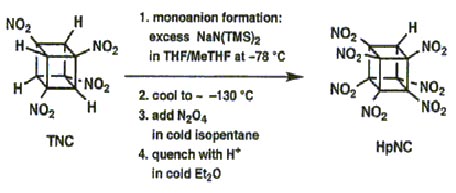
HpNC
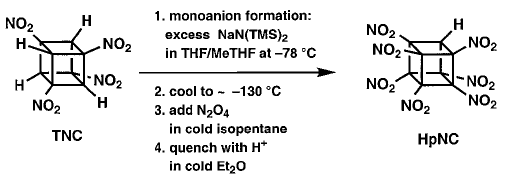
In this procedure TNC was treated with at least 4 equivalents of the base NaN(TMS)2 (where TMS = trimethylsilyl) at ±78 �C in 1:1 THF/MeTHF. After the mono sodium salt had formed, the solution was cooled to between ±125 and ±130°�C giving a clear, but very viscous fluid. This was stirred vigorously as excess N2O4 in cold isopentane was added. After one minute, the base was quenched, and the whole mixture was added to water. This resulted reproducibly in almost complete conversion of TNC (1 g scale) to HpNC (95% by NMR), isolated crystalline in 74% yield!
ONC
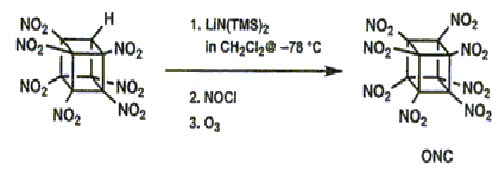
However, even in the presence of excess nitrating agent (N2O4 or many others) no indication
of any formation of ONC was ever seen. It is suspected that anion nitration with N2O4 proceeds by oxidation of the carbanion to the corresponding radical.Perhaps the anion of HpNC is too stabilized for this to occur. (HpNC is significantly ionized in neutral methanol.) This concept led to the use of the more powerful oxidant nitrosyl chloride. Addition of excess NOCl to a solution of the lithium salt of HpNC in dichloromethane at 78° �C followed by ozonation at 78° �C gave the long-sought ONC in 45±55% isolated yield on millimole scale. The intermediate product prior to oxidation is thought to be nitrosoheptanitrocubane.
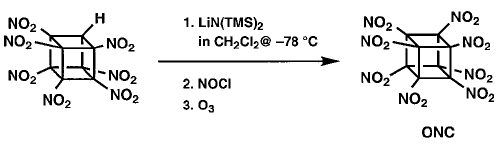
Finally, the magic molecule, the so called the impossible molecule, octanitrocubane was synthesised. But, how good are they and how useful are they? Let us discuss about it in the following section.
Properties of nitrocubane:
Neither HpNC nor ONC is detonated by hammer blows!
Both have decomposition points well above 200 �C. Octanitrocubane
sublimes unchanged at atmospheric pressure at 200 �C. HpNC forms beautiful, colorless, solvent-free crystals when
its solution in fuming nitric acid is diluted with sulphuric acid. Single-
crystal X-ray analysis confirmed the assigned structure and
provided an accurate density at 21 �C of 2.028 g cm±3, impressively
high for a C, H, N, O compound. Although octanitrocubane
catches the imagination with its symmetry, heptanitrocubane
currently is significantly easier to make than ONC. It is
denser, and it may be a more powerful, shock-insensitive explosive
than any now in use. According to page 41 of a 2004 IUPAC guide, cubane is the “preferred IUPAC name.”
- ‘ ‘Cubaneand Thomas W. Cole. Philip E. Eaton and Thomas W. Cole J. Am. Chem. Soc.; 1964; 86(15) pp 3157 – 3158; doi:10.1021/ja01069a041.
- The Cubane System Philip E. Eaton and Thomas W. Cole J. Am. Chem. Soc.; 1964; 86(5) pp 962 – 964; doi:10.1021/ja01059a072
- Michael B. Smith, Jerry March, March’s Advanced Organic Chemistry, 5 th Ed., John Wiley & Sons, Inc., 2001, p. 1459. ISBN 0-471-58589-0
- K. Kindler, K. Lührs, Chem. Ber., vol. 99, 1966, p. 227.
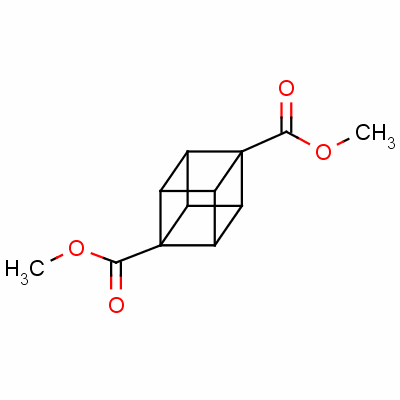
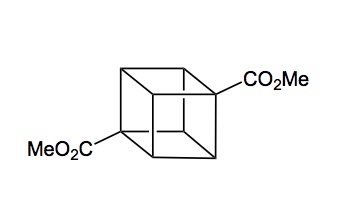
Dimethyl cubane-1,4-dicarboxylate
dimethyl 1,4-cubanedicarboxylate;
1,4-cubanedicarboxylic acid dimethyl*ester;
methyl 4-(methoxycarbonyl)pentacyclo[4.2.0.0<2,5>.0<3,8>.0<4,7>]octanecarboxylate
Pentacyclo(4.2.0.0(2,5).0(3,8).0(4,7))octane-1,4-dicarboxylic acid dimethyl ester
CAS 29412-62-2
| Molecular Weight: |
220.2213 |
| Molecular Formula: |
C12H12O4 |
| Density: |
1.684g/cm3 |
| Boiling Point(℃): |
270°C at 760 mmHg |
| Flash Point(℃): |
131.3°C |
| refractive_index: |
1.704 |
An interesting OPRD paper on the scale up of dimethyl cubane -1,4-dicarboxylate.

The work appeared in Organic Process Research and development, 2013, doi.org/10.1021/op400181g . It was carried out by an Australian group, John Tsanaktsidis, Michael Falkiner, Stuart Littler, Kenneth McRae and Paul Savage from CSIROand features a large-scale photochemical reaction which is very unusual to see in a scaled chemical process.
Extending their previous work from 1997, they scaled the following reaction.
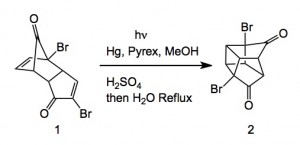
As is the norm with such reactions the reaction requires high dilution to be successful. In this case they used a tailor made photochemical reactor. A solution of 1 in methanol/water was pumped through the reactor at 4 L/minute and the conversion of 1 to 2 was noted as 1g/4 minutes of irradiation.
This meant a total time of 173 hours. Further processing of 2 through the double Favourskii ring contraction required significant development but eventually delivered the di-sodium salt corresponding to the di-ester of cubane.
One needs to be careful with these cubanes as they are, due to the highly strained nature of the system quite energetic materials, the do-acid and ester being more stable than the parent hydrocarbon. However the energy released upon warming above the melting point is not insignificant.
This paper represents a good demonstration of the scale-up of several very difficult chemical reactions, including excellent descriptive paragraphs of the problems and solutions. They are to be congratulated on a very nice piece of work.
See below
A scalable process for the preparation of high purity dimethyl 1,4-cubanedicarboxylate (3) is reported.
The work described herein builds on previous synthetic work from this and other laboratories, to provide a reliable process that can be used to prepare multigram quantities of 3 in a partially telescoped, 8 step process, with minimal purification of intermediates.
CSIRO Materials Science & Engineering, Ian Wark Laboratory, Bayview Avenue, Clayton Victoria 3168,Australia
Org. Process Res. Dev., 2013, 17 (12), pp 1503–1509
DOI: 10.1021/op400181g
Publication Date (Web): November 8, 2013

Scheme 5. Pilot-Scale Synthesis of Dimethyl 1,4-Cubanedicarboxylate (3)

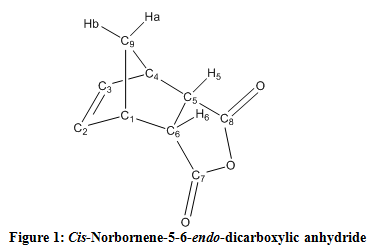
Figure 1. Cubane nucleus.
Step 5
A dry 100 L glass reactor was charged with the crude diacid 2 (1287 g), dry methanol (36 L), and Dowex ion-exchange resin 50WX8–100 (176 g) that was prewashed with 1 L of methanol. This mixture was then stirred (150 rpm), and heated under reflux for 18 h under an atmosphere of nitrogen. The mixture was then cooled to room temperature and filtered to remove the resin. The methanol solution mixture was then evaporated to dryness using a rotary evaporator (45 °C at 45 mmHg) leaving behind the crude diester 3 (863 g) as a dark brown solid. Purification by sublimation (100–120 °C/0.01 mmHg), followed by recrystallization from acetonitrile furnished the diester 3 (560 g, 30%), as a colorless solid,
mp 164.5 °C (lit. 161–162 °C).
(47) 1H NMR δ: 3.7, s, 6H 4.24, s, 6H, ring protons.
13C NMR δ: 47.03, 51.55, 55.77, 171.89.
………………………………..
http://cst-www.nrl.navy.mil/lattice/struk/c8h8.html.
http://www.ch.ic.ac.uk/local/projects/b_muir/Enter.html.
http://www.sciencedirect.com.
http://www.sciencenews.org/.
http://www.winbmdo.com/.
Bashir-Hashemi, A., New developments in cubane chemistry: phenylcubanes.
J. Am. Chem. Soc.;1988;110(21);7234-7235, 110(21), 7234-7235.
D.S.Calvao, p. m. v. b. B. A. C. J. a., Theooretical Characterization of oligocubane.
Synthetic Metals 102 (1999) 1410.
E. W. Della, E. F. M., H. K. Patney,Gerald L. Jones,; Miller, a. F. A.,
Vibrational Spectra of Cubane and Four
of Its Deuterated Derivatives.
Journal of the American Chemical Society / 101.25 / December 5, I979,7441-7457.
Galasso, V., Theoretical study of spectroscopic properties of cubane.
Chemical Physics 184 (1994) 107-114.
Kirill A. Lukin, J. L., Philip E. Eaton,*,Nobuhiro Kanomata,Juirgen Hain,Eric Punzalan,and
Richard Gilardi, Synthesis and Chemistry of 1,3,5,7-Tetranitrocubane Including
Measurement of Its Acidity, Formation of o-Nitro Anions, and
the First Preparations of Pentanitrocubane and Hexanitrocubane.
J. Am. Chem. Soc., Vol. 119, No. 41, 1997,9592-9602.
P.E.Eaton, Cubanes: starting Materials For the chemistry of 1990s and the New Century.
J. Am. Chem. Soc.;1992;31;1421-1436, 31, 1421-1436.
Philip E. Eaton, t. Y. X., t and Richard Gilardi*, Systematic Substitution on the Cubane Nucleus.
Synthesis and
Properties of 1,3,5-Trinitrocubane and 1,3,5,7-Tetranitrocubane
. J. Am. Chem. SOC.1993,115, 10195-10202.
Philip E. Eaton, R. L. G.; Zhang, a. M.-X., Polynitrocubanes: Advanced High-Density,
High-Energy Materials**. Adv. Mater. 2000, 12, No. 15, August 2.
Philip E. Eaton, Cubane: Starting Materials for the chemistry of the 1990s and the new century.
Angew.Chem.Int.Ed.Engl.1992,31,1421-1436.
Philip E. Eaton, t. Y. X., t and Richard Gilardi*, Systematic Substitution on the Cubane Nucleus.
Synthesis and
Properties of 1,3,5-Trinitrocubane and 1,3,5,7-Tetranitrocubane.
J. Am. Chem. SOC., Vol. 115, No. 22, 1993,10196-10202.
T. YILDIRIM, P. M. G., D. A. NEUMANN, P. E. EATONC and ‘T. EMRICK’, SOLID
CUBANE: A BRIEF REVIEW. Carbon Vol. 36, No. 5-6, pp. 809-815,1998.
Zhang, P. E. E. a. M.-X., Octanitrocubane: A New Nitrocarbon.
Propellants, Explosives, Pyrotechnics 27, 1 – 6 (2002).













 MAKE IN INDIA
MAKE IN INDIA









 amcrasto@gmail.com
amcrasto@gmail.com
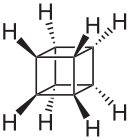 Cubane
Cubane


















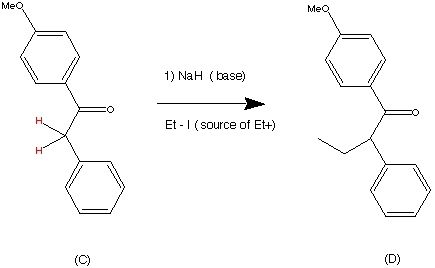
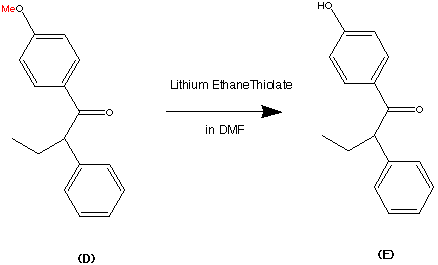
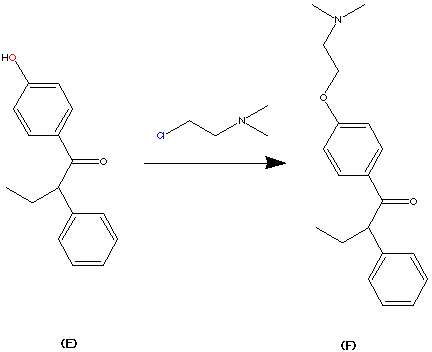
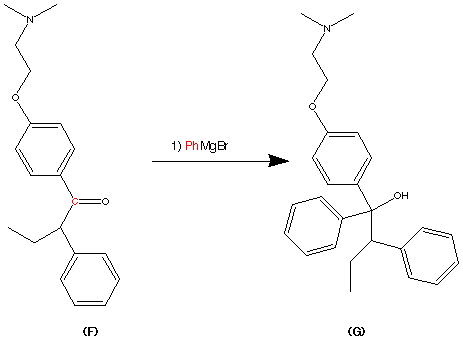
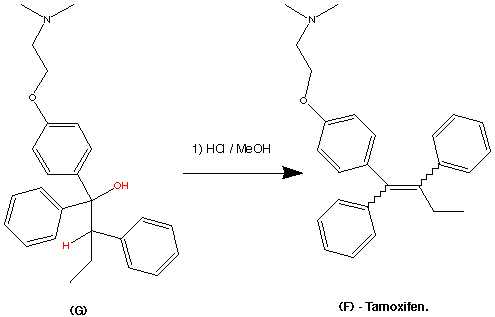
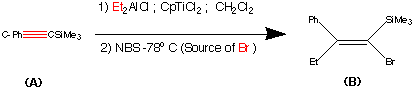
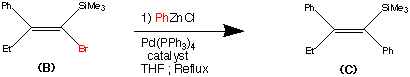
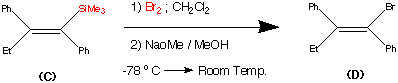











 99.9% purity is described. A simple, and safe process is developed for the recovery of tributyltin chloride from the tributyltin hydroxide, byproduct formed in the tetrazole ring construction, and reused in the synthesis of valsartan.
99.9% purity is described. A simple, and safe process is developed for the recovery of tributyltin chloride from the tributyltin hydroxide, byproduct formed in the tetrazole ring construction, and reused in the synthesis of valsartan.