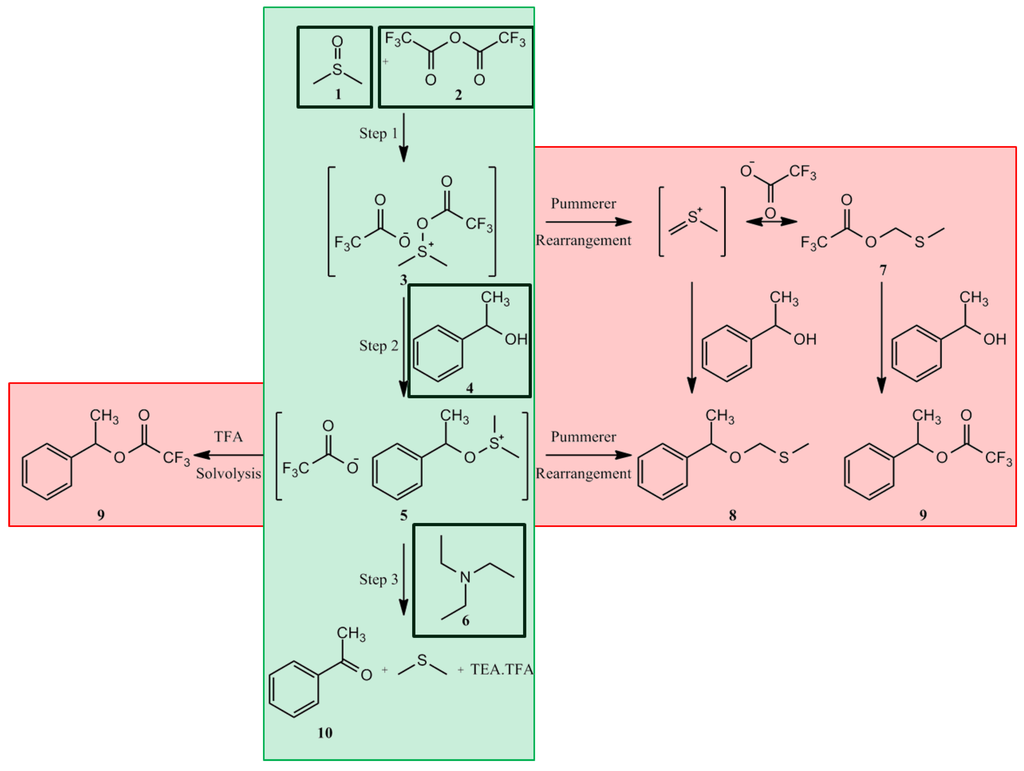
WO2015124764
ERREGIERRE S.P.A. [IT/IT]; Via Francesco Baracca, 19 I-24060 San Paolo D’argon (IT)
Erregierre SpA
DABIGATRAN ETEXILATE MESYLATE, INTERMEDIATES OF THE PROCESS AND NOVEL POLYMORPH OF DABIGATRAN ETEXILATE”
Abstract
A novel process is described for the production of Dabigatran etexilate mesylate, a 5 compound having the following structural formula: and two novel intermediates of said process.
(WO2015124764) SYNTHESIS PROCESS OF DABIGATRAN ETEXILATE MESYLATE, INTERMEDIATES OF THE PROCESS AND NOVEL POLYMORPH OF DABIGATRAN ETEXILATE click herefor patent
Dabigatran etexilate mesylate is an active substance developed by Boehringer
Ingelheim and marketed under the name Pradaxa® in the form of tablets for oral administration; Dabigatran etexilate mesylate acts as direct inhibitor of thrombin (Factor I la) and is used as an anticoagulant, for example, for preventing strokes in patients with atrial fibrillation or blood clots in the veins (deep vein thrombosis) that could form following surgery.
Dabigatran etexilate mesylate is the INN name of the compound 3-({2-[(4-{Amino-[(E)-hexyloxycarbonylimino]-methyl}-phenylamino)-methyl]-1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate methanesulphonate, having the following structural formula:

The family of compounds to which Dabigatran etexilate belongs was described for the first time in patent US 6,087,380, which also reports possible synthesis pathways.
The preparation of polymorphs of Dabigatran etexilate or Dabigatran etexilate mesylate is described in patent applications US 2006/0276513 A1 , WO 2012/027543 A1 , WO 2008/059029 A2, WO 2013/124385 A2, WO 2013/124749 A1 , WO 2013/1 1 1 163 A2 and WO 2013/144903 A1 , while patent applications WO 2012/044595 A1 , US 2006/0247278 A1 , US 2009/0042948 A2, US 2010/0087488 A1 and WO 2012/077136 A2 describe salts of these compounds.
One of the objects of the invention is to provide an alternative process for the preparation of Dabigatran etexilate mesylate and two novel intermediates of the process.
These objects are achieved with the present invention, which, in a first aspect thereof, relates to a process for the production of Dabigatran etexilate mesylate, comprising the following steps:
a) reacting 4-methylamino-3-nitrobenzoic acid (I) with thionyl chloride to give 4- methylamino-3-nitrobenzoyl chloride hydrochloride (II):

(I) (ID
b) reacting compound (II) with 3-(2-pyridylamino) ethyl propanoate (III) to give the compound 3-[(4-methylamino-3-nitro-benzoyl)-pyridyn-2-yl-amino]-ethyl propanoate (IV):

(II) (IV)
reducing compound (IV) with hydrogen to 3-[(3-amino-4-methyl benzoyl)-pyridin-2-yl-amino]ethyl propanoate (V):

(IV) (V)
d) reacting N-(4-cyanophenyl)glycine (VI) with 1 ,1 -carbonyldiimidazole (CDI) to give 4-(2-imidazol-1 -yl-2-oxo-ethylamino)-benzonitrile (VII):

(VI) (VII)
e) reacting compound (VII) with compound (V) obtained in step c) to give one of compounds 3-({3-[2-(4-cyano-phenylamino)-acetylamino]-4-methylamino- benzoyl}-pyridin-2-yl-amino)-ethyl propanoate (VIII) and 3-[(3-amino-4-{[(2- (4-cyano-phenylamino)-acetyl]-methylamino}-benzoyl)-pyridin-2-yl- amino]ethyl propanoate (IX), or a mixture of the two compounds (VIII) and (IX):

f) transforming, through treatment with acetic acid, compounds (VIII) or (IX) or the mixture thereof into the compound 3-({2-[(4-cyano-phenylamino)-methyl]- 1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate (X), and then treating compound (X) with hydrochloric or nitric acid to form the corresponding salt (XI):
CHsCOOH
[(VIII) ; (IX)]


wherein A is a chlorine or nitrate anion;
liberating in solution compound (X) from salt (XI), and reacting compound (X) in solution with ethyl alcohol in the presence of hydrochloric acid and 2,2,2-trifluoroethanol to give the compound 3-({2-[(4-ethoxycarbonimidoyl-phenylamino)-methyl]-1 -methyl-1 H-benzimidazol-5-carbonyl}-pyridin-2-yl-amino)-ethyl propanoate hydrochloride (XII):

reacting compound (XII) with ammonium carbonate to form compound Dabigatran ethyl ester (XIII):

reacting compound (XIII) with maleic acid to produce the maleate salt thereof (XI 11 ‘) and isolating the latter:

j) reacting maleate salt (XI 11 ‘) with hexyl chloroformate to give compound Dabigatran etexilate (XIV :
hexyl chloroformate

k) reacting compound (XIV) with methanesulfonic acid to give the salt Dabigatran etexilate mesylate:

a gatran etex ate mesy ate
EXAMPLE 12
Preparation of Dabigatran etexilate mesylate (step k).
All the Dabigatran etexilate obtained in Example 1 1 (4.7 kg; 7.49 moles) is loaded into a reactor along with 28.2 kg of acetone and the mass is heated at 50-60 °C until a complete solution is obtained; it is then filtered to remove suspended impurities. The filtered solution is brought to 28-32 °C. Separately, a second solution is prepared by dissolving 0.705 kg (7.34 moles) of methanesulfonic acid in 4.7 kg of acetone; the second solution is cooled down to 0-10 °C. The second solution is poured into the Dabigatran etexilate solution during 30 minutes, while maintaining the temperature of the resulting solution at 28-32 °C with cooling. The salt of the title is formed. The mass is maintained at 28-32 °C for 2 hours, then cooled to 18-23 °C to complete precipitation and the system is maintained at this temperature for 2 hours; lastly, centrifugation takes place, washing the precipitate with 5 kg of acetone. The precipitate is dried at 60 °C.
4.88 kg of Dabigatran etexilate mesylate, equal to 6.74 moles of compound, are obtained, with a yield in this step of 90%.
EXAMPLE 13
0.5 g of the crystalline compound (XIV) obtained in Example 1 1 are ground thoroughly and loaded into the sample holder of a Rigaku Miniflex diffractometer with copper anode.
The diffractogram shown in Figure 1 is obtained; a comparison with the XRPD data of the known Dabigatran etexilate polymorphs allows to verify that the polymorph of Example 1 1 is novel.
EXAMPLE 14
0.7 g of the crystalline compound (XIV) obtained in Example 1 1 are loaded into
the sample holder of a Perkin-Elmer DSC 6 calorimeter, performing a scan from ambient T to 350 °C at a rate of 10 °C/min in nitrogen atmosphere. The graph of the test is shown in Figure 2, and shows three endothermic phenomena with peaks at 83.0-85.0 °C, 104.0-104.2 °C and 129.9 °C; events linked to the thermal decomposition of the compound are evident at about 200 °C.

Figure 1 is an XRPD spectrum of the novel polymorph of Dabigatran etexilate of the invention;

Figure 2 is the graph of a DSC test on the novel polymorph of Dabigatran etexilate of the invention.
ERREGIERRE S.p.A

Pietro Carlo Gargani, CEO and president of ERREGIERRE S.p.A., oversees a company with a firm commitment to serving its customers innovative products

ERREGIERRE was founded by two entrepreneurs in 1974 in San Paolo d’Argon, in the northern Italian region of Bergamo. It lodged one of its first major …


///////////ERREGIERRE S.p.A, DABIGATRAN, WO 2015124764
































 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..


















 LIONEL MY SON
LIONEL MY SON