Tout sur les médicaments הכל על תרופות كل شيئ عن الأدوية Все о наркотиках 关于药品的一切 డ్రగ్స్ గురించి అన్ని 마약에 관한 모든 것 Όλα για τα Ναρκωτικά Complete Tracking of Drugs Across the World by Dr Anthony Melvin Crasto, Worldpeacepeaker, worlddrugtracker, PH.D (ICT), MUMBAI, INDIA, Worlddrugtracker, Helping millions, 9 million hits on google on all websites, 2.5 lakh connections on all networks, “ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
A multistep sequential flow synthesis of isopropyl phenol is demonstrated, involving 4-step exothermic, endothermic, and temperature sensitive reactions such as nitration, reduction, diazotization, and high temperature hydrolysis. Nitration of cumene with fuming nitric acid produces 2- and 4-nitrocumene which are converted into respective cumidines by the hydrogenation using Pd/Ni catalyst in H-cube with gravity separation. Hydrolysis of in situ generated diazonium salts in the boiling acidic conditions is carried out using integration of flow and microwave-assisted synthesis. 58% of 4-isopropyl phenol was obtained. The sequential flow synthesis can be applied to synthesize other organic compounds involving this specific sequence of reactions.
Telescoped Sequence of Exothermic and Endothermic Reactions in Multistep Flow Synthesis
Dr.Amol A. Kulkarni
Chemical Engineering & Process Development
CSIR-National Chemical Laboratory
Dr. Amol A. Kulkarni is a Scientist in the Chemical Engineering Division at the National Chemical Laboratory. He did his B. Chem. Eng. (1998), M. Chem. Eng (2000) and Ph.D. in chemical engineering (2003) all from the University Dept. of Chem. Technology (UDCT, Mumbai). In 2004 he worked at the Max Planck Institute-Magdeburg (Germany) as a Alexander von Humboldt Research Fellow. At NCL he is driving a research program on the design of microreactors and exploring their applications for continuous syntheses including of nanoparticles. He has been awarded with the Max-Planck-Visiting Fellowship from the Max-Planck-Society, Munich for 2008-2011. His research areas include: (i) design and applications of microreactors, (ii) design of multiphase reactors, (iii) experimental and computational fluid dynamics, and (iv) nonlinear dynamics of coupled systems. He is an active member of Initiative for Research and Innovation in Science (IRIS) supported by Intel’s Education Initiative to organize National Science Fair and popularize science in India.
Yachita Sharma is a PhD student at CSIR-National Chemical Laboratory, Pune (India). She received her MSc in Applied Organic Chemistry in 2010. Her work focuses on exploring the continuous flow synthesis involving exothermic reactions and their integration.
Arun was born and raised in Koregaon, Maharashtra, India. He completed his bachelors and masters in chemical sciences from Shivaji Unversity, Kolhapur, India. Currently, He is pursuing his Ph. D. under the supervision of Dr. Amol A. Kulkarni and Dr. B. L. V. Prasad. His work mainly focuses on flow synthesis of nanoparticles, drug formulation, and polymers. He develops new synthesis procedures and translates into flow chemistry to increase productivity with excellent control over the quality of the product. He is also exploring the application of nanoparticles in catalysis, electronics and pharmaceutical fields. He specializes in microwave-assisted continuous flow synthesis of nanomaterial and organic intermediate. Apart from his research, he actively pursues Yoga and spirituality. He also likes to play volleyball and has competed in inter CSIR tournaments.
/////////isopropyl phenol, flow chem,
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
FLOW CHEMISTRY, flow synthesisComments Off on Development and Scale-Up of a Continuous Reaction for Production of an Active Pharmaceutical Ingredient Intermediate
Flow reactor equipment that was used in the piloting of the aldol flow chemistry. (a) The tube-in-shell heat exchangers were used to control stream temperature upstream and downstream of the (b) Y-mixer. Valves and ports on Y-mixer enabled flushing of lines and incorporation of inline thermocouples.
Examples of continuous flow reactions in the laboratory setting are becoming commonplace in pharmaceutical drug substance research. Developing these processes for robust commercialization and identifying the scale-up parameters remains a challenge. An aldol reaction in the formation of an active pharmaceutical ingredient intermediate was developed in flow at the milliliter scale. Research focused on identifying conditions that led to robust and stable operating conditions. Desired reaction performance was achieved in various mixers across reactor scales by identifying conditions that led to similar flow regimes. Conditions from the lab were transferred to the pilot plant to successfully process ∼200 kg of the starting material.
Development and Scale-Up of a Continuous Reaction for Production of an Active Pharmaceutical Ingredient Intermediate
A flow chemistry process for sustainable operations was developed by utilizing THF as a cosolvent to improve the solubility of the reagent degradants. Process robustness was further established by understanding the impact of mixing, residence time, and solvent composition on reaction performance. Identifying a suitable flow regime via the Reynolds number was identified as the scaling parameter for this flow reaction and used to scale the flow reaction from 20 mL/min in the lab to 1.6 L/min in the production environment. A modular flow reactor skid was fabricated for facile integration of flow chemistry components with existing batch equipment and was used to process 200 kg of starting material.
We report mixing characterization of five lab-scale and eight production-scale static mixers using a modified fourth Bourne reaction. An efficient inline method relying on UV–vis spectroscopy was developed to streamline analysis of the product distribution. As a result of these studies, we have designed, 3D-printed, and characterized a stainless steel static mixer. This approach enabled the evaluation of different configurations and ensured efficient scale-up across development and commercial facilities that should allow for enhanced portability of mixing-sensitive processes.
Advancing Flow Chemistry Portability: A Simplified Approach to Scaling Up Flow Chemistry
FLOW CHEMISTRY, flow synthesisComments Off on Utilization of fluoroform for difluoromethylation in continuous flow: a concise synthesis of α-difluoromethyl-amino acids
Manuel Kockinger, Tanja Ciaglia, Michael Bersier, Paul Hanselmann, Bernhard Gutmann, C. Oliver Kappe
Difluoromethylated esters, malonates and amino acids (including the drug eflornithine) are obtained by a gas-liquid continuous flow protocol employing the abundant waste product fluoroform as an atom-efficient reagent.
Utilization of fluoroform for difluoromethylation in continuous flow: a concise synthesis of α-difluoromethyl-amino acids
b Microreactor Technology, Lonza AG, CH-3930 Visp, Switzerland
c Research Center Pharmaceutical Engineering GmbH (RCPE), Inffeldgasse 13, 8010 Graz, Austria
Abstract
Fluoroform (CHF3) can be considered as an ideal reagent for difluoromethylation reactions. However, due to the low reactivity of fluoroform, only very few applications have been reported so far. Herein we report a continuous flow difluoromethylation protocol on sp3 carbons employing fluoroform as a reagent. The protocol is applicable for the direct Cα-difluoromethylation of protected α-amino acids, and enables a highly atom efficient synthesis of the active pharmaceutical ingredient eflornithine.
Methyl 3,3-(difluoro)-2,2-diphenylpropanoate (2a) The product mixtures were collected and the solvent removed in vacuo. The products were isolated by thin layer chromatography (dichloromethane/hexane = 3/2 (v/v)). Yield: 173 mg (0.62 mmol, 62%); 93% by 19F NMR ;light yellow viscous liquid. 1 H NMR (300 MHz, D2O): δ = 7.45 – 7.19 (m, 10H), 6.90 (t, 2 JHF = 55.0 Hz, 1H), 3.79 (s, 3H). 13C NMR (75 MHz, D2O): δ = 171.1, 136.3, 129.8, 128.3, 128.2, 115.6 (t, 1 JCF = 246.2 Hz), 64.7, 53.1.19F NMR (282 MHz, D2O):δ = -123.0 (d, 2 JHF = 55.0 Hz).
Conclusions
A gas–liquid continuous flow difluoromethylation protocol employing fluoroform as a reagent was reported. Fluoroform, a by-product of Teflon manufacture with little current synthetic value, is the most attractive reagent for difluoromethylation reactions. The continuous flow process allows this reaction to be performed within reaction times of 20 min with 2 equiv. of base and 3 equiv. of fluoroform. Importantly, the protocol allows the direct Cα-difluoromethylation of protected α-amino acids. These compounds are highly selective and potent inhibitors of pyridoxal phosphate-dependent decarboxylases. The starting materials are conveniently derived from the commercially available α-amino acid methyl esters, and the final products are obtained in excellent purities and yields after simple hydrolysis and precipitation. The developed process appears to be especially appealing for industrial applications, where atom economy, sustainability, reagent cost and reagent availability are important factors.
FLOW CHEMISTRY, flow synthesis, UncategorizedComments Off on Contribution of microreactor technology and flow chemistry to the development of green and sustainable synthesis
Department of Pharmacy – Drug Sciences, University of Bari “A. Moro”, FLAME-Lab – Flow Chemistry and Microreactor Technology Laboratory, Via E. Orabona 4, 70125, Bari. Italy
Guest Editor: L. Vaccaro Beilstein J. Org. Chem.2017,13, 520–542. doi:10.3762/bjoc.13.51 Received 14 Nov 2016, Accepted 20 Feb 2017, Published 14 Mar 2017
Abstract
Microreactor technology and flow chemistry could play an important role in the development of green and sustainable synthetic processes. In this review, some recent relevant examples in the field of flash chemistry, catalysis, hazardous chemistry and continuous flow processing are described. Selected examples highlight the role that flow chemistry could play in the near future for a sustainable development.
Green chemistry’s birth was driven by the necessity to consider and face the urgent question of sustainability. Chemical production concerns an extended range of fields such as textiles, construction, food, cosmetic components, pharmaceuticals and so forth. An innovative approach to the chemistry world requires new strategies and criteria for an intelligent chemistry. It is understood that all this matter has big implications in economy and politics. Recent studies predicted a growth of green chemical processing up to $100 billion in 2020 (Pike Research study) [1]. All this offers important and arduous challenges expressed in terms of new synthetic strategies using sustainable, safe, and less toxic materials. On green chemistry we can read Paul Anastas and John Warne’s 12 principles, set up in 1998, which illustrate the characteristics of a greener chemical process or product [2]. Microreactor technology and flow chemistry could play a pivotal role in the context of sustainable development. In fact, flow chemistry is becoming a new technique for fulfilling several of the twelve green chemistry principles. The microreactor approach, could provide protection, preserves atom economy, guarantees less hazardous chemical synthesis and allows the use of safer solvents and auxiliaries. Furthermore, it pushes towards designing of chemistry with a lower environmental and economic impact, enhance the importance of catalysis, allows real-time analysis for pollution prevention and provides inherently safer chemistry (Figure 1) [3]. Without claiming to be exhaustive, in this review we report recently published representative synthetic applications that demonstrate the growing contribution of flow chemistry and microreactor technology in green and sustainable synthesis [4-7].
Figure 1: Microreactor technologies and flow chemistry for a sustainable chemistry.
Review
Flow microreactors: main features
The peculiar properties of microreactors [8] derive from their small size and can be ascribed mainly to the following characteristics: a) fast mixing: in a flow microreactor, in striking contrast to batch conditions, mixing takes place by molecular diffusion so that a concentration gradient can be avoided; b) high surface-to-volume ratio: the microstructure of microreactors allows for a very rapid heat transfer enabling fast cooling, heating and, hence, precise temperature control; c) residence time: it is the period of time the solution of reactants spend inside the reactor, and it gives a measure of the reaction time. The residence time is strictly dependent on the characteristics of the reactor (i.e., length of the channels, volume), and on the flow rate. The residence time is one of the crucial factors to be considered in optimizing flow reactions, especially when unstable or short-lived reactive intermediates are concerned. Microreactor technology provides also several benefits. Safety benefits, because of the high efficiency in heat exchange, and avoided accumulation of unstable intermediates. Economy benefits, due to lower manufacturing and operating costs, reduced work-up procedures, use of less raw materials and solvents and reduced waste. Chemistry benefits associated to the use of microreactor technology are the improved yields and selectivities, the possibility to conduct reactions difficult or even impossible to perform in batch, and the use of reaction conditions that allow exploring new chemical windows [9].
Contribution of flash chemistry to green and sustainable synthesis
The concept of flash chemistry as a “field of chemical synthesis using flow microreactors where extremely fast reactions are conducted in a highly controlled manner to produce desired compounds with high selectivity” was firstly introduced by Yoshida [10]. Flash chemistry can be considered a new concept in both organic and sustainable synthesis involving chemical transformations that are very difficult or practically impossible to conduct using conventional batch conditions. With the aim to show how flow microreactor technology and flash chemistry could contribute to the development of a sustainable organic synthesis, very recent examples have been selected and will be discussed here. In the context of green chemistry [11], protecting-group free organic synthesis has received particular attention in the last years, because of atom economy [12-15] and reduction of synthetic steps [16]. It has been demonstrated by Yoshida that protecting-group-free synthesis could be feasible using flash chemistry and microreactor technology [17,18]. Recently, Yoshida and co-workers developed flash methods for the generation of highly unstable carbamoyl anions, such as carbamoyllithium, using a flow microreactor system [19]. In particular, they reported that starting from different substituted carbamoyl chloride 1 and lithium naphthalenide (LiNp) it was possible to generate the corresponding carbamoyllithium 2, that upon trapping with different electrophiles provided several amides and ketoamide 3(Scheme 1).
Scheme 1: A flow microreactor system for the generation and trapping of highly unstable carbamoyllithium species.
The use of an integrated microflow system allowed the preparation of functionalized α-ketoamides by a three-component reaction between carbamoyllithium, methyl chloroformate and organolithium compounds bearing sensitive functional groups (i.e., NO2, COOR, epoxide, carbonyl) (Scheme 2).
Scheme 2: Flow synthesis of functionalized α-ketoamides.
It should be stressed that this kind of sequential transformations are practically impossible to perform using conventional batch chemistry because of the incompatibility of sensitive functional groups with organolithiums, and because of the high chemical and thermal instability of the intermediates.
In 2015 Yoshida reported another remarkable finding on the use of protecting-group-free organolithium chemistry. In particular, the flash chemistry approach was exploited for generating benzyllithiums bearing aldehyde or ketone carbonyl groups [20]. This reaction could be problematic for two reasons: a) the competing Wurtz-type coupling, (i.e., the coupling of benzyllithiums with the starting benzyl halides); b) the nucleophilic attack of organolithium species to aldehyde or ketone carbonyl groups (Scheme 3).
Scheme 3: Reactions of benzyllithiums.
The authors reported that the extremely fast micromixing avoided undesired Wurtz-type coupling [21,22]. It is well known, that competitive reactions can be controlled or even avoided under fast micromixing [23-27]. Moreover, high-resolution residence time control was essential for survival of carbonyl groups. In fact, this transformation can be achieved only with a residence time of 1.3 ms at −78 °C. Under these flow conditions, the aldehyde or ketone carbonyl moiety can survive the nucleophilic organolithium attack. Remarkably, the flow microreactor system allowed also the generation of benzyllithiums at 20 °C, rather than under cryogenic (−95 °C) conditions adopted with a conventional batch protocol. In addition, THF could be used in place of mixed solvents (Et2O/THF/light petroleum). Under the optimized conditions, the reactions of benzyllithiums with different electrophiles, gave adduct products in good yields (Scheme 4).
Scheme 4: Trapping of benzyllithiums bearing carbonyl groups enabled by a flow microreactor. (Adapted with permission from [18], copyright 2015 The Royal Society of Chemistry).
Another useful aspect of the flash chemistry relies on the possibility to generate highly reactive intermediates, such as halomethyllithium carbenoids, that need to be used under internal-quenching technique in batch mode. In 2014, the first example of effective external trapping of a reactive chloromethyllithium (CML) has been reported [28]. α-Haloalkyllithiums are a useful class of organometallic reagents widely employed in synthetic chemistry. In fact, they allow the direct homologation of carbonyl compounds and imines leading to β-halo-alcohols and amines that are useful building blocks [29-31]. This work represents a remarkable example of flash chemistry, and has elements of sustainability considering that in batch macroreactors, in order to avoid metal-assisted α-elimination, in situ quenching, an excess of reagents, and very low temperature are required [32,33].
Running the reaction in a flow system at −40 °C, by using residence times between 0.18–0.31 s high yields of homologated products have been obtained under external quenching conditions (Scheme 5).
Scheme 5: External trapping of chloromethyllithium in a flow microreactor system.
The results described above nicely show the potential, as green technology, of flow microreactor systems for synthetic processes involving highly unstable intermediates. Another nice example on the use of microreactor technology for the development of sustainable chemical processes, is represented by the direct introduction of the tert-butoxycarbonyl group into organometallic reagents [34]. The reaction between organolithium reagents and di-tert-butyl dicarbonate run under flow conditions, allowed a straightforward preparation of several tert-butyl esters. The use of a flow process resulted more efficient, versatile and sustainable compared to batch. Moreover, this operationally simple procedure complements well with the already available strategies for the preparation of tert-butyl esters, avoiding the use of inflammable and explosive gaseous isobutylene [35], the use of harsh conditions [36], the use of peroxides [37], the use of toxic gas such as CO or transition metals [38-42]. The flow process, for the direct C-tert-butoxycarbonylation of organolithiums, has been optimized in a green solvent such as 2-MeTHF by a precise control of the residence time, and without using cryogenic conditions (Scheme 6). In addition, many organolithiums were generated from the corresponding halo compounds by a halogen/lithium exchange reaction using hexyllithium as a more sustainable base [43,44].
Scheme 6: Scope for the direct tert-butoxycarbonylation using a flow microreactor system.
The concept of flash chemistry has been successfully employed for outpacing fast isomerization reactions. The accurate control of the residence time, realized in a microreactor, could suppress or avoid isomerization of unstable intermediates. This is often unavoidable when the same reactions are run in batch mode [45-47].
Yoshida and Kim recently provided an astonishing example on the potential of flash chemistry in controlling fast isomerization of organolithiums [48]. The authors designed a chip microreactor (CMR), able to deliver a reaction time in the range of submilliseconds (0.33 ms) under cryogenic conditions. By using such an incredible short residence time, it was possible to overtake the very rapid anionic Fries rearrangement, and chemoselectively functionalize ortho-lithiated aryl carbamates (Scheme 7).
Scheme 7: Control of anionic Fries rearrangement reactions by using submillisecond residence time. (Adapted with permission [43], copyright 2016 American Association for the Advancement of Science).
This CMR has been developed choosing a fluoroethylene propylene–polymide film hybrid for fabrication because this material offers exceptional physical toughness at low temperature and high pressure as well as chemical inertness. The most relevant aspect of this microreactor, concerns the 3D design of the mixing zone (Figure 2). The mixing efficiency was evaluated on the basis of computational fluids dynamics (CFD). The simulation results showed that serpentine 3D-structured channels (Figure 2), possessing five turns after each mixing point in a total length of 1 mm, was able to deliver the highest mixing efficiency. The inner volume for the reactor was of 25 μL. This CMR provides mixing efficiency levels of 95% with a total flow rates of 7.5 mL/min corresponding to a residence time of about 0.3 milliseconds.
Figure 2: Chip microreactor (CMR) fabricated with six layers of polyimide films. (Reproduced with permission from [43], copyright 2016 American Association for the Advancement of Science).
To show the potential use of this microdevice in organic synthesis, the synthesis of Afesal [49], a biologically active compound having anthelmintic activity was reported as application.
This outstanding result by Yoshida and Kim, demonstrates how microdevices and flash chemistry could contribute to the development of new sustainable synthetic strategies, and how microreactor technology could help in taming the reactivity of unstable species [50].
Contribution of continuous-flow metal-, organo-, and photocatalysis in green chemistry
The development of continuous-flow catalysis is appealing because it combines the advantages of a catalytic reaction with the benefits of flow microreactors. Under homogeneous conditions a soluble catalyst, which flows through the reactor together with the reactants, is employed. At the end of the process, a separation step would be required in order to remove the catalyst and byproducts. On the other hand, heterogeneous catalysis is widely used in the synthesis of bulk and fine chemicals. In a continuous-flow process, the catalyst can be fixed on a suitable hardware, and the reaction mixture allowed to flow through the system. The use of recyclable catalysts in continuous-flow conditions represents an innovative strategy for the development of more environmentally friendly synthesis. In the last decade, organic photochemistry got a sort of renaissance, emerging as useful approach in modern sustainable and green synthesis.
Concerning the heterogeneous catalysis with palladium, practical procedures for recovering and reusing of the catalysts have been recently reported [51-53]. A versatile Pd-catalysed synthesis of polyfunctionalized biaryls, using a flow microreactor, has been recently reported by Yoshida [54]. Using the integrated microflow system reported in Scheme 8, arylboronic esters were prepared by a lithiation/borylation sequence, and used in a Suzuki–Miyaura coupling in a monolithic reactor. A remarkable aspect of the process was the use of an integrated supported monolithic Pd(0) catalyst that allowed to perform cross-coupling reactions in continuous flow mode (Scheme 8).
Scheme 8: Flow microreactor system for lithiation, borylation, Suzuki–Miyaura coupling and selected examples of products.
This integrated microflow system allow to handle the borylation of aryl halides (Ar1X), and the subsequent Suzuki–Miyaura coupling using different aryl halide (Ar2X). Without requiring the protection of sensitive functionalities, running the flow system using a residence time (tR) of about 4.7 min at a temperature above 100 °C, high yields of coupling products were obtained. Noteworthy, the Suzuki–Miyaura coupling did not require the use of a base. The authors applied the presented method to the synthesis of adapalene, used in the treatment of acne, psoriasis, and photoaging.
Fluorinated aromatic compounds are extremely important in agrochemical, pharmaceutical and medicinal fields [55-58]. Buchwald and co-workers suggested a telescoped homocatalysis procedure consisting of a three-step sequence (metalation, zincation and Negishi cross-coupling) which furnishes an easy access to a variety of functionalized 2-fluorobiaryl and heteroaryl products (Scheme 9) [59]. This strategy is rightfully considered green because it guarantees the employment of readily available and cheap starting materials, the safe handling of highly thermally unstable or dangerous intermediates, and the use of higher temperature with respect to the batch mode in which the proposed reactions have to be carried out at −78 °C.
Scheme 9: Experimental setup for the flow synthesis of 2-fluorobi(hetero)aryls by directed lithiation, zincation, and Negishi cross-coupling. (Adapted with permission from [53], copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim).
The use of 2-MeTHF as greener solvent, contributes to further validate the green procedure. The 2-MeTHF solutions of fluoroarenes 4 together with the hexane solution of n-BuLi were pumped into the flow system at −40 °C. The generated organozinc intermediate meets the solution of haloarenes and the catalyst, leading to the formation of the desired products 5a–j (Scheme 9). Noteworthy, the homogeneous catalysis requires only 1% of the XPhos-based palladium catalyst. A sonication bath was employed to prevent clogging and the reaction required a residence time of 15 min.
Next, they turned their attention to the arylation of fluoro-substituted pyridines. The regioselective lithiation of halopyridines with lithium diisopropylamide (LDA) was conducted under mild conditions on substrate 6(Scheme 10). The addition of a little amount of THF was necessary in order to avoid clogging and the tendency of the lithiated intermediate to eliminate.
Scheme 10: Experimental setup for the coupling of fluoro-substituted pyridines. (Adapted with permission from [53], copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim).
The optimized conditions were suitable for the functionalization of 2-fluoropyridine, 2,6- difluoropyridine and 4-(trifluoromethyl)pyridine leading to products 7a–g reported in Scheme 10. Another promising field is the sustainable flow organocatalysis, and recently Pericàs reported an interesting synthesis and application of a recyclable immobilized analogue of benzotetramisole (BMT) used in a catalytic enantioselective Michael addition/cyclization reactions under continuous-flow conditions (Scheme 11) [60].
Scheme 11: Continuous flow process setup for the preparation of 11 (Reproduced with permission from [54], copyright 2015 American Chemical Society).
Resin-bound catalyst 10 was swollen with dichloromethane in a medium-pressure chromatography column used as a reactor. Dichloromethane solutions of substrate 9 reacted with the mixed phenylacetic pivalic anhydride (deriving from phenylacetic acetic (8) and pivaloyl chloride) inside the catalytic reactor producing the expected products 11. This ingenious system was equipped with an in-line FTIR probe, for monitoring the transformation, and an in line liquid–liquid separator to avoid tedious work-up procedures, thus saving solvents, resources and optimizing work times. This system was demonstrated to work for 11 h with higher conversion and enantioselectivity (er >99.9%) in comparison to the batch mode [61]. Pericàs and co-workers taking advantage of the high catalytic activity, robustness and recyclability of the supported catalyst, performed also straightforward gram synthesis of target compounds.
In the context of photocatalysis and oxidations using flow microreactors [62,63], Noël reported a metal-free photocatalytic aerobic oxidation of thiols to disulfides under continuous-flow conditions [64]. Disulfides are useful molecules employed as drugs, anti-oxidants or pesticides as well as rubber vulcanizating agents [65]. Symmetric disulfides are generally obtained by oxidative coupling of thiols [66]. Noël and co-workers set up a microflow system equipped with a mass flow controller (MFC) able to introduce pure oxygen as the oxidant to oxidize a solution of thiol containing 1% of Eosin Y. The flow stream was exposed to white LED light in order to activate the reaction, and a dilution with pure EtOH was needed at the output to avoid clogging (Scheme 12). The residence time of 20 min guaranteed a limited irradiation time and high purity of the products.
Scheme 12: Continuous-flow photocatalytic oxidation of thiols to disulfides.
The disulfides were obtained with excellent yields, and the process was executed on challenging thiols as in the case of disulfide 12 (Scheme 12), used as food flavour additive [67]. To demonstrate the usefulness of the flow methodology, and its applicability, the photocatalytic aerobic oxidation of a peptide to obtain oxytocin in continuous flow was reported (Scheme 12). Full conversion was achieved in water with 200 s of residence time.
Noël optimized, for the first time, a trifluoromethylation of aromatic heterocycles by continuous-flow photoredox catalysis. The process benefited from the use of microreactor technology and readily available photocatalysts. The process was also employable for perfluoroalkylation. The developed process occurred in less time with respect to batch mode, and under milder conditions. The set-up of the reactor allowed for the use of gaseous CF3I by means of a mass flow controller. Selected examples of trifluoroalkylated products are reported in Scheme 13[68].
Scheme 13: Trifluoromethylation by continuous-flow photoredox catalysis.
Tranmer reported a “traceless reagents” chemistry with the continuous-flow photosynthesis of 6(5H)-phenanthridinones, poly(ADP-ribose) polymerase (PARP) inhibitors [69]. The relevance of the work resides in the use of green solvents, the absence of heavy metals, the use of convenient temperatures, and the increased safety by eliminating UV-exposure locating the UV lamp within the microreactor. Hazard of fires caused by the hot UV lamps approaching the auto-ignition temperature of flammable solvents, very often underestimated, is totally prevented thanks to a specific cooling system. 2-Halo-N-arylbenzamides were converted into 6(5H)-phenanthridinones by a photocyclization reaction. In order to run this step, a flow system with a photochemical reactor equipped with a medium pressure Hg lamp and 10 mL reactor coil, was employed. Good yields were obtained from different 2-chlorobenzamides disclosing that either electron-donating or electron withdrawing ortho-substituents were tolerated (Scheme 14).
Scheme 14: Flow photochemical synthesis of 6(5H)-phenanthridiones from 2-chlorobenzamides.
A metal- and catalyst-free arylation procedure carried out under continuous-flow conditions was recently reported by Fagnoni [70]. This photochemical process allowed for the preparation of a wide range of synthetic targets by Ar–Csp3, Ar–Csp2 and Ar–Csp bond-forming reactions. The use of a photochemical flow reactor, consisting of a polyfluorinated tube reactor wrapped around a 500 W Hg lamp, allowed to overcome batch limitations paving the way for metal-free arylation reactions via phenyl cations. Derivatives 14a–g were prepared with this greener flow approach (Scheme 15) starting from mesitylene 13, and haloarenes using short irradiation times (<6 h), and a 5:1 MeCN/H2O mixture.
Scheme 15: Synthesis of biaryls 14a–g under photochemical flow conditions.
The reported results show how photochemistry hold the potential to become a green tool for the development of sustainable photochemical flow synthesis.
Hazardous chemistry by using green and sustainable continuous-flow microreactors
We have already shown how continuous-flow technology could play an important role in improving chemical processes [5,71], providing different advantages over traditional batch mode. However, the hazardous nature of some chemicals makes handling at conventional lab or industrial scale difficult. The use of microreactors and continuous-flow chemistry offers the possibility to perform reactions using dangerous or hazardous materials that cannot be used in batch mode. In other word, syntheses previously “forbidden” for safety reasons, such as those involving diazo compounds, hydrazine, azides, phosgene, cyanides and other hazardous chemicals could be performed with relatively low risk using flow technology [72-76].
Several research groups investigated this aspect, as highlighted by several available reviews [77,78]. Here we describe very recent reports with the aim to highlight the potential of flow chemistry in the field of hazardous chemistry under a greener perspective.
Diazo compounds are recognized as versatile reagents in organic synthesis. Nevertheless, diazo compounds are also considered highly energetic reagents [79,80]. For this reason, the in situ generation of such reagents has been investigated under flow conditions. Moody and co-workers reported a new method for the in situ generation of diazo compounds as precursors of highly reactive metal carbenes (Scheme 16) [81].
Scheme 16: Flow oxidation of hydrazones to diazo compounds.
As reported in Scheme 16, diazo species 18 could be generated from simple carbonyls 15 and hydrazine (16). Intermediate hydrazones 17 can be converted into the corresponding diazo compounds by oxidation using a recyclable oxidant based on N-iodo-p-toluenesulfonamide potassium salt. The possibility to regenerate a functionalized resin by simple washing with aqueous KI3/KOH solution makes the process more sustainable. This method produces KI solution as waste, and it is an alternative way for the direct oxidation of hydrazones, that often requires the use of heavy metals such as HgO, Pb(OAc)4 and AgO [82,83].
The diazo compounds could be collected as solution in dichloromethane at the output of the flow system, and obtained sufficiently pure for further use without requiring handling or isolation. Further mixing of solutions containing diazo derivatives to a solution containing a Rh(II) catalyst, and reactants such as amines, alcohols or aldehydes led to a wide range of products as reported in Scheme 17.
Scheme 17: Synthetic use of flow-generated diazo compounds.
Ley’s group developed several continuous-flow approaches for generating diazo species from hydrazones [84,85]. Under flow conditions, diazo compounds were reacted with boronic acids in order to generate reactive allylic and benzylic boronic acids further employed for iterative C–C bond forming reactions [86]. The generation of unstable diazo species was possible using a cheap, recyclable and less toxic oxidant, MnO2. The flow stream was accurately monitored by in-line FTIR spectroscopy in order to maximize the formation of the diazo compound (Scheme 18) [87].
Scheme 18: Ley’s flow approach for the generation of diazo compounds.
Starting from this initial investigation, Ley and co-workers developed an elegant application of this strategy for a sequential formation of up to three C–C bonds in sequence, by an iterative trapping of boronic acid species. The sequence starts with the reaction of diazo compound 20, generated under flow conditions, and boronic acid 19 (Scheme 19). Further sequential coupling with diazo compounds 21 and 22 led to boronates 23 or protodeboronated products 24 at the end of the sequence (Scheme 19).
Scheme 19: Iterative strategy for the sequential coupling of diazo compounds.
With the aim to exploit the versatility of this approach, Ley and co-workers reported the allylations of carbonyl electrophiles such as aldehydes using the above reported strategy for the generation of allylboronic acids. The flow protocol considers the reaction of diazo compounds 25 (generated in flow) with boronic acid 26 and aldehyde 27 (Scheme 20). By this new iterative coupling it was possible to obtain alcohols as products. The usefulness of the method was demonstrated with the preparation in good yield (60%) of a precursor of the natural product bakuchiol 28 (Scheme 20) [88].
Scheme 20: Integrated synthesis of Bakuchiol precursor via flow-generated diazo compounds.
The microreactor technology offers the advantage to handle hazardous components such as hydrazine and molecular oxygen, which represent alternative reagents for selective reduction of C=C double bonds. In fact, combination of hydrazine hydrate (N2H4·H2O) and O2 provide diimide (HN=NH) as reducing agent. Nevertheless, this strategy is rarely used in traditional batch chemistry for safety reason. Kappe and co-workers recently developed a reduction of the alkene to the corresponding alkane, by a catalyst-free generation of diimide by oxidation of hydrazine monohydrate (N2H4·H2O) with molecular oxygen [89,90]. The flow system set-up is reported in Scheme 21, and consists in a HPLC pump for delivering the alkene and hydrazine monohydrate, while O2 was delivered by a mass-flow controller (MFC) from a standard compressed-gas cylinder. After combination of the reagent streams, the resulting segmented flow was pumped through a heated residence unit (RTU) consisting in a fluorinated tube with low gas permeability (Scheme 21).
Scheme 21: Kappe’s continuous-flow reduction of olefines with diimide.
The flow system reported in Scheme 21 was able to reduce alkenes with high yields and selectivity by using residence times in the range of 10 to 30 min at 100 °C, and by employing a slight excess of hydrazine. Importantly, this strategy is compatible with sensitive functional groups such as silyl ether, halogenes, and benzyl groups. A very nice application of this approach was the highly selective reduction of artemisinic acid to dihydroartemisinic acid, which are of interest in the synthesis of the antimalarial drug artemisinin. This industrially relevant reduction was executed by using O2 at 20 bar, four residence units at 60 °C and consecutive feedings with N2H4·H2O in order to obtain full conversion in dihydroartemisinic acid (29, DHAA, Scheme 22).
Scheme 22: Multi-injection setup for the reduction of artemisinic acid.
Continuous-flow sustainable production of APIs
With the aim to demonstrate the potential of microreactor technology and flow chemistry in sustainable synthesis, recent outstanding “proof of concepts” will be described. Kobayashi and co-workers reported a multistep continuous-flow synthesis of a drug target via heterogeneous catalysis. The developed process not requiring any isolation of intermediates, separation of the catalyst or other work-up procedures can be considered sustainable [91]. The syntheses of (S)-rolipram and a γ-aminobutyric acid (GABA) derivative were accomplished. Readily available starting materials and columns containing chiral heterogeneous catalysts to produce enantioenriched materials were employed. It is worth mentioning that this work represents a very nice example on the use of chiral catalysis in a multistep flow synthesis of a drug target on gram scale. The multistep synthesis of (S)-rolipram reported in Scheme 23 begins from a benzaldehyde derivative which undergoes a Henry-type reaction with nitromethane in the first flow step (Flow I). The resulting nitroalkene undergoes an asymmetric addition catalyzed by a supported PS–(S)-pybox–calcium chloride catalyst at 0 °C using two columns (Flow II). This is the enantio-determining step of the process. The stereochemistry of the adduct can be simply switched to the opposite enantiomer, by using the enantiomeric supported catalyst PS–(R)-pybox–calcium chloride. The enantiomeric excess of the products was about 96%. Two more steps consisting in a Pd-catalyzed hydrogenation reaction and a decarboxylation (Flow III and Flow IV) led to the target (S)-rolipram in 50% overall yield. The systems was designed in order to keep the level of the palladium in solution as low as possible (<0.01 ppm).
Scheme 23: Flow reactor system for multistep synthesis of (S)-rolipram. Pumps are labelled a, b, c, d and e; Labels A, B, C, D, E and F are flow lines. X are molecular sieves; Y is Amberlyst 15Dry; Z is Celite. (Reproduced with permission from [84], copyright 2015 Nature Publishing Group).
Another outstanding proof of concept, which demonstrates the potential of flow chemistry for sustainable pharmaceutical manufacturing, has been recently reported by Jensen and his research team. The research team set up a compact and reconfigurable manufacturing platform for the continuous-flow synthesis and formulation of active pharmaceutical ingredients (APIs) [92]. The “mini” plant (reported in Figure 3) was very compact in size [1.0 m × 0.7 m × 1.8 m, (W × L × H)], and low-weighing (about 100 kg) and was able to perform complex multistep synthesis, work-up procedures as well as purification operations such as crystallization. This platform was also equipped with devices for real-time monitoring and final formulation of high purity APIs. For the preparation of target molecules, commercially available starting materials were employed. The platform was tested for the production and supply of hundreds to thousands doses per day of diphenhydramine hydrochloride, lidocaine hydrochloride, diazepam and fluoxetine hydrochloride.
Remarkably, for future applications of the platform, the produced medicines also met the U.S. Pharmacopeia standards.
Figure 3: Reconfigurable modules and flowcharts for API synthesis. (Reproduced with permission from [85], copyright 2016 American Association for the Advancement of Science).
The future use of this kind of platform would concern the “on-demand” production or the “instantaneous” production of short-lived pharmaceuticals (Figure 4). Other advantageous concerns of this reconfigurable platform are the lower production costs, the higher safety, the automation (computer controlled processes), the reduced waste (production could be done where is needed and in the right amount).
Figure 4: Reconfigurable system for continuous production and formulation of APIs. (Reproduced with permission from [85], copyright 2016 American Association for the Advancement of Science).
Conclusion
Flow chemistry and manufacturing engineering have become largely acknowledged as viable and very often superior alternative to batch processing. Continuous-flow techniques offer increased safety, scalability, reproducibility, automation, reduced waste and costs, and accessibility to a wide range of new chemical possibilities, seldom not accessible through classic batch chemistry. All those benefits are even more noteworthy and outstanding than what they might seem, because they widely fulfil most of the green chemistry principles. In this short overview, we tried to highlight progresses and potential of flow chemistry in the field of sustainable synthesis. Thus, it is expected that flow chemistry and microreactor technology could deeply change the way to perform sustainable chemical production in the near future [93].
Newman, S. G.; Jensen, K. F. Green Chem.2013,15, 1456–1472. doi:10.1039/c3gc40374b
Return to citation in text: [1]
Professor Jun-ichi Yoshida firstly introduced the concept of “micro + flow = green” during a plenary lecture at the 14th IMRET conference held in Beijing (China) in Septembeer 2016.
Return to citation in text: [1]
Yoshida, J.-i.; Kim, H.; Nagaki, A. ChemSusChem2011,4, 331–340. doi:10.1002/cssc.201000271
Return to citation in text: [1]
Hessel, V.; Schouten, J. C.; Renken, A.; Wang, Y.; Yoshida, J.-i., Eds. Handbook of Micro Reactors; Wiley-VCH: Weinheim, 2009.
Return to citation in text: [1]
Yoshida, J.-i. Flash Chemistry: Fast Organic Synthesis in Microsystems; Wiley-VCH: Weinheim, 2008. doi:10.1002/9780470723425
Return to citation in text: [1]
Poliakoff, M.; Fitzpatrick, J. M.; Ferren, T. R.; Anastas, P. T. Science2002,297, 807–810. doi:10.1126/science.297.5582.807
Return to citation in text: [1]
Trost, B. M. Acc. Chem. Res.2002,35, 695–705. doi:10.1021/ar010068z
Return to citation in text: [1]
Wender, P. A.; Verma, V. A.; Paxton, T. J.; Pillow, T. H. Acc. Chem. Res.2008,41, 40–49. doi:10.1021/ar700155p
Return to citation in text: [1]
Kim, H.; Nagaki, A.; Yoshida, J.-i. Nat. Commun.2011,2, No. 264. doi:10.1038/ncomms1264
Return to citation in text: [1]
Nagaki, A.; Yoshida, J.-i. Microreactor Technology in Lithium Chemistry, in: Lithium Compounds in Organic Synthesis from Fundamentals to Applications. Luisi, R.; Capriati, V., Eds.; Wiley-VCH: Weinheim, 2014; pp 491–512. doi:10.1002/9783527667512.ch17
Return to citation in text: [1] [2]
Nagaki, A.; Takahashi, Y.; Yoshida, J.-i. Angew. Chem., Int. Ed.2016,55, 5327–5331. doi:10.1002/anie.201601386
Return to citation in text: [1]
Nagaki, A.; Tsuchihashi, Y.; Haraki, S.; Yoshida, J.-i. Org. Biomol. Chem.2015,13, 7140–7145. doi:10.1039/C5OB00958H
Return to citation in text: [1]
Rys, P. Acc. Chem. Res.1976,10, 345–351. doi:10.1021/ar50106a001
Return to citation in text: [1]
Rys, P. Angew. Chem., Int. Ed. Engl.1977,16, 807–817. doi:10.1002/anie.197708073
Return to citation in text: [1]
Nagaki, A.; Togai, M.; Suga, S.; Aoki, N.; Mae, K.; Yoshida, J.-i. J. Am. Chem. Soc.2005,127, 11666–11675. doi:10.1021/ja0527424
Return to citation in text: [1]
Yoshida, J.-i. Basics of Flow Microreactor Synthesis; SpringerBriefs in Molecular Science; Springer: Tokyo, 2015. doi:10.1007/978-4-431-55513-1
Return to citation in text: [1]
Yoshida, J.-i.; Nagaki, A.; Iwasaki, T.; Suga, S. Chem. Eng. Technol.2005,28, 259–266. doi:10.1002/ceat.200407127
Return to citation in text: [1]
Nagaki, A.; Takabayashi, N.; Tomida, Y.; Yoshida, J.-i. Org. Lett.2008,18, 3937–3940. doi:10.1021/ol8015572
Return to citation in text: [1]
Nagaki, A.; Ichinari, D.; Yoshida, J.-i. Chem. Commun.2013,49, 3242–3244. doi:10.1039/c3cc40392k
Return to citation in text: [1]
Degennaro, L.; Fanelli, F.; Giovine, A.; Luisi, R. Adv. Synth. Catal.2015,357, 21–27. doi:10.1002/adsc.201400747
Return to citation in text: [1]
Pace, V.; Holzer, W.; De Kimpe, R. Chem. Rec.2016,16, 2061–2076. doi:10.1002/tcr.201600011
Return to citation in text: [1]
Gessner, V. H. Chem. Commun.2016,52, 12011–12023. doi:10.1039/C6CC05524A
Return to citation in text: [1]
Pace, V. Aust. J. Chem.2014,67, 311–313. doi:10.1071/CH13416
Return to citation in text: [1]
Emerson, C. R.; Zakharov, L. N.; Blakemore, P. R. Chem. – Eur. J.2013,19, 16342–16356. doi:10.1002/chem.201302511
See for a tactic to prolong the life-time of carbenoids consisting in the introduction of anion-stabilizing groups and see references cited therein.
Return to citation in text: [1]
Kupper, C.; Molitor, S.; Gessner, V. H. Organometallics2014,33, 347–353. doi:10.1021/om4010862
See for examples of “stabilized” chloro carbenoids stable at room temperature and references cited therein.
Return to citation in text: [1]
Degennaro, L.; Maggiulli, D.; Carlucci, C.; Fanelli, F.; Romanazzi, G.; Luisi, R. Chem. Commun.2016,52,9554–9557. doi:10.1039/C6CC04588J
Return to citation in text: [1]
Furniss, B. S.; Hannaford, A. J.; Smith, P. W. G.; Tatchell, A. R. Vogel’s Textbook of Practical Organic Chemistry, 5th ed.; Wiley: New York, 1989; p 1266.
Return to citation in text: [1]
Dawar, P.; Raju, M. B.; Ramakrishna, R. A. Tetrahedron Lett.2011,52, 4262–4265. doi:10.1016/j.tetlet.2011.04.100
Return to citation in text: [1]
Zhu, Y.; Wei, Y. RSC Adv.2013,3, 13668–13670. doi:10.1039/c3ra40246k
Return to citation in text: [1]
Zhang, H.; Shi, R.; Ding, A.; Lu, L.; Chen, B.; Lei, A. Angew. Chem., Int. Ed.2012,51, 12542–12545. doi:10.1002/anie.201206518
Return to citation in text: [1]
Majek, M.; Jacobi von Wangelin, A. Angew. Chem., Int. Ed.2015,54, 2270–2274. doi:10.1002/anie.201408516
Return to citation in text: [1]
Magano, J.; Dunetz, J. R. Chem. Rev.2011,111, 2177–2250. doi:10.1021/cr100346g
Return to citation in text: [1]
Brennführer, A.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed.2009,48, 4114–4133. doi:10.1002/anie.200900013
Return to citation in text: [1]
Xin, Z.; Gøgsig, T. M.; Lindhardt, A. T.; Skrydstrup, T. Org. Lett.2012,14, 284–287. doi:10.1021/ol203057w
Return to citation in text: [1]
Zenzola, M.; Degennaro, L.; Trinchera, P.; Carroccia, L.; Giovine, A.; Romanazzi, G.; Mastrorilli, P.; Rizzi, R.; Pisano, L.; Luisi, R. Chem. – Eur. J.2014,20, 12190–12200. doi:10.1002/chem.201403141
Return to citation in text: [1] [2] [3]
Parisi, G.; Capitanelli, E.; Pierro, A.; Romanazzi, G.; Clarkson, G. J.; Degennaro, L.; Luisi, R. Chem. Commun.2015,51, 15588–15591. doi:10.1039/C5CC06323J
Return to citation in text: [1]
Giovine, A.; Musio, B.; Degennaro, L.; Falcicchio, A.; Nagaki, A.; Yoshida, J.-i.; Luisi, R. Chem. – Eur. J.2013,19, 1872–1876. doi:10.1002/chem.201203533
Return to citation in text: [1]
Tomida, Y.; Nagaki, A.; Yoshida, J.-i. J. Am. Chem. Soc.2011,133, 3744–3747. doi:10.1021/ja110898s
Return to citation in text: [1]
Nagaki, A.; Matsuo, C.; Kim, S.; Saito, K.; Miyazaki, A.; Yoshida, J.-i. Angew. Chem., Int. Ed.2012,51,3245–3248. doi:10.1002/anie.201108932
Return to citation in text: [1]
Kim, H.; Min, K.-I.; Inoue, K.; Im, D. J.; Kim, D.-P.; Yoshida, J.-i. Science2016,352, 691–694. doi:10.1126/science.aaf1389
Return to citation in text: [1]
Semple, J. E.; Rossignol, J.-F. Pharmaceutical compositions and methods of use of salicylanilides for treatment of hepatitis viruses. PCT Int. Appl. WO2012058378 A1, May 3, 2012.
Return to citation in text: [1]
Degennaro, L.; Carlucci, C.; De Angelis, S.; Luisi, R. J. Flow Chem.2016,6, 136–166. doi:10.1556/1846.2016.00014
Return to citation in text: [1]
Pavia, C.; Ballerini, E.; Bivona, L. A.; Giacalone, F.; Aprile, C.; Vaccaro, L.; Gruttadauria, M. Adv. Synth. Catal.2013,355, 2007–2018. doi:10.1002/adsc.201300215
Return to citation in text: [1]
de M. Muñoz, J.; Alcázar, J.; de la Hoz, A.; Díaz-Ortiz, A. Adv. Synth. Catal.2012,354, 3456–3460. doi:10.1002/adsc.201200678
Return to citation in text: [1]
Mennecke, K.; Sodolenko, W.; Kirschning, A. Synthesis2008, 1589–1599. doi:10.1055/s-2008-1072579
Return to citation in text: [1] [2] [3]
Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed.; Wiley-VCH: Weinheim, 2013. doi:10.1002/9783527651351
Return to citation in text: [1]
Wang, J.; Sànchez-Rosellò, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev.2014,114, 2432–2506. doi:10.1021/cr4002879
Return to citation in text: [1]
Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem.2015,58, 8315–8359. doi:10.1021/acs.jmedchem.5b00258
Return to citation in text: [1]
Roesner, S.; Buchwald, S. L. Angew. Chem., Int. Ed.2016,55, 10463–10467. doi:10.1002/anie.201605584
Return to citation in text: [1]
Izquierdo, J.; Pericas, M. A. ACS Catal.2016,6, 348–356. doi:10.1021/acscatal.5b02121
Return to citation in text: [1]
Izquierdo, J.; Ayats, C.; Henseler, A. H.; Pericàs, M. A. Org. Biomol. Chem.2015,13, 4204–4209. doi:10.1039/C5OB00325C
Return to citation in text: [1]
Cambié, D.; Bottecchia, C.; Straathof, N. J. W.; Hessel, V.; Noël, T. Chem. Rev.2016,116, 10276–10341. doi:10.1021/acs.chemrev.5b00707
Return to citation in text: [1]
Gemoets, H. P. L.; Su, Y.; Shang, M.; Hessel, V.; Luque, R.; Noël, T. Chem. Soc. Rev.2016,45, 83–117. doi:10.1039/C5CS00447K
Return to citation in text: [1]
Talla, A.; Driessen, B.; Straathof, N. J. W.; Milroy, L.-G.; Brunsveld, L.; Hessel, V.; Noël, T. Adv. Synth. Catal.2015,357, 2180–2186. doi:10.1002/adsc.201401010
Return to citation in text: [1]
Cremlyn, R. J. An Introduction to Organosulfur Chemistry; Wiley–VCH: New York, 1996.
Return to citation in text: [1]
Blank, I.; Pascual, E. C.; Devaud, S.; Fay, L. B.; Stadler, R. H.; Yeretzian, C.; Goodman, B. A. J. Agric. Food Chem.2002,50, 2356–2364. doi:10.1021/jf011329m
Return to citation in text: [1]
Straathof, N. J. W.; Gemoets, H. P. L.; Wang, X.; Schouten, J. C.; Hessel, V.; Noël, T. ChemSusChem2014,7, 1612–1617. doi:10.1002/cssc.201301282
Return to citation in text: [1]
Fang, Y.; Tranmer, G. K. Med. Chem. Commun.2016,7, 720–724. doi:10.1039/C5MD00552C
Return to citation in text: [1]
Bergami, M.; Protti, S.; Ravelli, D.; Fagnoni, M. Adv. Synth. Catal.2016,358, 1164–1172. doi:10.1002/adsc.201600019
Return to citation in text: [1]
Wiles, C.; Watts, P. Green Chem.2012,14, 38–54. doi:10.1039/C1GC16022B
Return to citation in text: [1]
Yoshida, J.-i. Flash Chemistry: Fast Organic Synthesis in Microsystems; John Wiley & Sons: Chichester, UK, 2008. doi:10.1002/9780470723425
Return to citation in text: [1]
Yoshida, J.-i. Chem. Commun.2005, 4509–4516. doi:10.1039/b508341a
Return to citation in text: [1]
Yoshida, J.-i.; Nagaki, A.; Yamada, T. Chem. – Eur. J.2008,14, 7450–7459. doi:10.1002/chem.200800582
Return to citation in text: [1]
Nieuwland, P. J.; Koch, K.; van Harskamp, N.; Wehrens, R.; van Hest, J. C. M.; Rutjes, F. P. J. T. Chem. – Asian J.2010,5, 799–805. doi:10.1002/asia.200900705
Return to citation in text: [1]
Gutmann, B.; Cantillo, D.; Kappe, C. O. Angew. Chem., Int. Ed.2015,54, 6688–6728. doi:10.1002/anie.201409318
Return to citation in text: [1]
Movsisyan, M.; Delbeke, E. I. P.; Berton, J. K. E. T.; Battilocchio, C.; Ley, S. V.; Stevens, C. V. Chem. Soc. Rev.2016,45, 4892–4928. doi:10.1039/C5CS00902B
Return to citation in text: [1]
Clark, J. D.; Shah, A. S.; Peterson, J. C.; Patelis, L.; Kersten, R. J. A.; Heemskerk, A. H.; Grogan, M.; Camden, S. Thermochim. Acta2002,386, 65–72. doi:10.1016/S0040-6031(01)00760-2
Return to citation in text: [1]
Hosmane, R. S.; Liebman, J. F. Struct. Chem.2002,13, 501–503. doi:10.1023/A:1020573723147
Return to citation in text: [1]
Nicolle, S. M.; Hayes, C. J.; Moody, C. J. Chem. – Eur. J.2015,21, 4576–4579. doi:10.1002/chem.201500118
Return to citation in text: [1]
Regitz, M.; Maas, G. Diazo Compounds Properties and Synthesis; Academic Press: Orlando, Florida, 1986.
Return to citation in text: [1]
Soldi, C.; Lamb, K. N.; Squitieri, R. A.; González-López, M.; Di Maso, M. J.; Shaw, J. T. J. Am. Chem. Soc.2014,136, 15142–15145. doi:10.1021/ja508586t
See for a recent example of hydrazone oxidation using manganese dioxide.
Return to citation in text: [1]
Roda, N. M.; Tran, D. N.; Battilocchio, C.; Labes, R.; Ingham, R. J.; Hawkins, J. M.; Ley, S. V. Org. Biomol. Chem.2015,13, 2550–2554. doi:10.1039/C5OB00019J
Return to citation in text: [1] [2]
Poh, J.-S.; Tran, D. N.; Battilocchio, C.; Hawkins, J. M.; Ley, S. V. Angew. Chem., Int. Ed.2015,54, 7920–7923. doi:10.1002/anie.201501538
Return to citation in text: [1] [2] [3]
Battilocchio, C.; Feist, F.; Hafner, A.; Simon, M.; Tran, D. N.; Allwood, D. M.; Blakemore, D. C.; Ley, S. V. Nat. Chem.2016,8, 360–367. doi:10.1038/nchem.2439
Return to citation in text: [1]
Tran, D. N.; Battilocchio, C.; Lou, S.-B.; Hawkins, J. M.; Ley, S. V. Chem. Sci.2015,6, 1120–1125. doi:10.1039/C4SC03072A
Return to citation in text: [1]
Esumi, T.; Yamamoto, C.; Fukuyama, Y. Synlett2013,24, 1845–1847. doi:10.1055/s-0033-1338968
Return to citation in text: [1]
Pieber, B.; Martinez, S. T.; Cantillo, D.; Kappe, C. O. Angew. Chem., Int. Ed.2013,125, 10431–10434. doi:10.1002/ange.201303528
Return to citation in text: [1]
Pieber, B.; Glasnov, T.; Kappe, C. O. Chem. – Eur. J.2015,21, 4368–4376. doi:10.1002/chem.201406439
Return to citation in text: [1]
Tsubogo, T.; Oyamada, H.; Kobayashi, S. Nature2015,520, 329–332. doi:10.1038/nature14343
Return to citation in text: [1]
Adamo, A.; Beingessner, R. L.; Behnam, M.; Chen, J.; Jamison, T. F.; Jensen, K. F.; Monbaliu, J.-C. M.; Myerson, A. S.; Revalor, E. M.; Snead, D. R.; Stelzer, T.; Weeranoppanant, N.; Wong, S. Y.; Zhang, P. Science2016,352, 61–67. doi:10.1126/science.aaf1337
Return to citation in text: [1]
Hessel, V.; Kralisch, D.; Kockmann, N., Eds. Novel Process Windows: Innovative Gates to Intensified and Sustainable Chemical Processes; Wiley-VCH: Weinheim, 2015.
Return to citation in text: [1]
How to cite this article:
Fanelli, F.; Parisi, G.; Degennaro, L.; Luisi, R. Beilstein J. Org. Chem.2017,13, 520–542. doi:10.3762/bjoc.13.51
As an organic chemist I’m involved in the development of new sustainable synthetic methodologies for the construction of new molecules with defined stereochemistry and functional properties.
Jointly with my coworkers we are involved in three main research themes:
1. Heterosubstituted Organolithiums. We mainly explore the reactivity of lithiated 3,4,5,6-membered N,S,O-heterocycles (aziridines, azetidines, oxazetidines, thietanes, oxazolines, piperazines, morfolines) and their utility in stereoselective synthesis. Our approach is focused in establishing the chemical and configurational stability of the lithiated intermediates as well as their structure in solution by using modern spectrometric and spectroscopic techniques such as in line -IR, in line-MS, NMR and DOSY.
2. Microreactor Technology and Flow-Chemistry. With the aim to design more sustainable synthetic processes, we set up, at the Depatment of Pharmacy, a well equipped “flow chemistry laboratory” named FLAME-Lab, for the development of continuous-flow microreactor-mediated organometallic and organocatalytic synthesis in both homegenous and heterogenous conditions.
3. Molecular Dynamics. As a “curiosity driven” research activity, we investigate the dynamic behavior of small molecules that could function as molecular switches with “on-off” states and as versatile scaffolds useful in catalysis and in “dynamic-controlled and predictable reactivity”.
Green Chem., 2017, Advance Article DOI: 10.1039/C7GC02261A, Communication
Shawn Parisien-Collette, Corentin Cruche, Xavier Abel-Snape, Shawn K. Collins
Polycyclic heterocycles can be formed in good to excellent yields via photochemical conversion of the corresponding substituted aryl azides under irradiation with purple LEDs in a continuous flow reactor.
Photochemical intramolecular amination for the synthesis of heterocycles
aShawn Parisien-Collette, Corentin Cruché, Xavier Abel-Snape and Prof, Dr Shawn K. Collins, Department of Chemistry and Centre in Green Chemistry and Catalysis, Université de Montréal, CP 6128 Station Downtown, Montréal, Canada H3C 3J7 E-mail:shawn.collins@umontreal.ca
Abstract
Polycyclic heterocycles can be formed in good to excellent yields via photochemical conversion of the corresponding substituted aryl azides under irradiation with purple LEDs in a continuous flow reactor. The experimental set-up is tolerant to UV-sensitive functional groups while affording diverse carbazoles, as well as an indole and pyrrole framework, in short reaction times. The photochemical method is presumed to progress through a mechanism differing from the other methods of azide activation involving transition metal catalysis.
Methyl 9H-carbazole-2-carboxylate (9): Following the Photodecomposition Procedure A, starting from Methyl 2’-azido-[1,1’-biphenyl]-4-carboxylate, the crude mixture was purified by silica gel column chromatography (100 % hexanes → 10 % ethyl acetate in hexanes), to afford the desired product as a white solid (24.3 mg, 72 % yield). Following the Photodecomposition Procedure B, starting from Methyl 2’-azido-[1,1’- biphenyl]-4-carboxylate, the crude mixture was purified by silica gel column chromatography (100 % hexanes → 10 % ethyl acetate in hexanes), to afford the desired product as a white solid (27.7 mg, 82 % yield). NMR data was in accordance with what was previously reported.16
16 Takamatsu, K.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2014, 16, 2892-2895
NEXT…………..
4-Isopropyl-9H-carbazole (14): Following the Photodecomposition Procedure A, starting from 2-azido-2’-isopropyl-1,1’-biphenyl, the crude mixture was purified by silica gel column chromatography (100 % hexanes → 10 % ethyl acetate in hexanes), to afford the desired product as a yellow solid (16.6 mg, 53 % yield). Following the Photodecomposition Procedure B, starting from 2-azido-2’-isopropyl-1,1’-biphenyl and using ethyl acetate as the solvant, the crude mixture was purified by silica gel column chromatography (100 % hexanes → 10 % ethyl acetate in hexanes), to afford the desired product as a yellow solid (16.0 mg, 51 % yield).
Revisiting the deoxydehydration of glycerol towards allyl alcohol under continuous-flow conditions
Green Chem., 2017, 19,3006-3013 DOI: 10.1039/C7GC00657H, Paper
Nelly Ntumba Tshibalonza, Jean-Christophe M. Monbaliu
Highly selective flash deoxydehydration of glycerol towards allyl alcohol under continuous-flow conditions.
aCenter for Integrated Technology and Organic Synthesis, Department of Chemistry, University of Liège, B-4000 Liège (Sart Tilman), Belgium E-mail:jc.monbaliu@ulg.ac.be
Abstract
The deoxydehydration (DODH) of glycerol towards allyl alcohol was revisited under continuous-flow conditions combining a microfluidic reactor setup and a unique reactive dynamic feed solution approach. Short reaction times, high yield and excellent selectivity were achieved at high temperature and moderate pressure in the presence of formic acid, triethyl orthoformate, or a combination of both. Triethyl orthoformate appeared as a superior reagent for the DODH of glycerol, with shorter reaction times, lower reaction temperatures and more robust conditions. In-line IR spectroscopy and computations provided different perspectives on the unique reactivity of glycerol O,O,O-orthoesters.
FLOW CHEMISTRY, flow synthesisComments Off on Continuous Microflow Synthesis of Fuel Precursors from Platform Molecules Catalyzed by 1,5,7-Triazabicyclo[4.4.0]dec-5-ene
The first continuous flow synthesis of C8–C16 alkane fuel precursors from biobased platform molecules is reported. TBD (1,5,7-triazabicyclo[4.4.0]dec-5-ene) was found to be a recyclable and highly efficient organic base catalyst for the aldol condensation of furfural with carbonyl compounds, and the selectivity of mono- or difuryl product can be easily regulated by adjusting the molar ratio of substrates. By means of flow technique, a shorter reaction time, satisfactory output, and continuous preparation are achieved under the present procedure, representing a significant advance over the corresponding batch reaction conditions.
Continuous Microflow Synthesis of Fuel Precursors from Platform Molecules Catalyzed by 1,5,7-Triazabicyclo[4.4.0]dec-5-ene
3-pentanone (100 mmol, 8.6 g) and furfural (100 mmol, 9.6 g) were diluted with MeOH-H2O to 40 mL in stream 1, catalyst TBD (10 mmol, 1.39 g) were diluted with MeOH-H2O (v/v = 1/1) to 40 mL in stream 2, the two streams was purged in a 0.2 mL/min speed into slit plate mixer and at the 353 K passed tubing reactor. Finally, the product was extracted with EtOAc and water, the obtained organic layer was evaporated and purified by silica gel flash chromatography (25:1 hexane-EtOAc) to provide the analytically pure product for further characterization, the aqueous phase was collected and reused.According to the general procedure afforded 14.92 g (91%) of product 1a, isolated as pale yellow oil;
FLOW CHEMISTRY, flow synthesis, phase 1Comments Off on Development of a concise, scalable synthesis of a CCR1 antagonist utilizing a continuous flow Curtius rearrangement
Green Chem., 2017, Advance Article DOI: 10.1039/C6GC03123D, Paper
Maurice A. Marsini, Frederic G. Buono, Jon C. Lorenz, Bing-Shiou Yang, Jonathan T. Reeves, Kanwar Sidhu, Max Sarvestani, Zhulin Tan, Yongda Zhang, Ning Li, Heewon Lee, Jason Brazzillo, Laurence J. Nummy, J. C. Chung, Irungu K. Luvaga, Bikshandarkoil A. Narayanan, Xudong Wei, Jinhua J. Song, Frank Roschangar, Nathan K. Yee, Chris H. Senanayake
A convergent and robust synthesis of a developmental CCR1 antagonist is described using continuous flow technology
A convergent, robust, and concise synthesis of a developmental CCR1 antagonist is described using continuous flow technology. In the first approach, following an expeditious SNAr sequence for cyclopropane introduction, a safe, continuous flow Curtius rearrangement was developed for the synthesis of a p-methoxybenzyl (PMB) carbamate. Based on kinetic studies, a highly efficient and green process comprising three chemical transformations (azide formation, rearrangement, and isocyanate trapping) was developed with a relatively short residence time and high material throughput (0.8 kg h−1, complete E-factor = ∼9) and was successfully executed on 40 kg scale. Moreover, mechanistic studies enabled the execution of a semi-continuous, tandem Curtius rearrangement and acid–isocyanate coupling to directly afford the final drug candidate in a single, protecting group-free operation. The resulting API synthesis is further determined to be extremely green (RPG = 166%) relative to the industrial average for molecules of similar complexity.
Development of a concise, scalable synthesis of a CCR1 antagonist utilizing a continuous flow Curtius rearrangement
aDepartment of Chemical Development, Boehringer Ingelheim Pharmaceuticals, Inc., 900 Ridgebury Road, Ridgefield, USA E-mail: maurice.marsini@boehringer-ingelheim.com
In September 2010, a randomized, double-blind, placebo-controlled, phase I study (NCT01195688; 1279.1; 2010-021187-15) was initiated in healthy male volunteers (expected n = 64) in Germany, to assess the safety, pharmacokinetics and pharmacodynamics of BI-638683. The study was completed in December 2010 . In June 2014, data were presented at the EULAR 2014 Annual Meeting in Paris, France. A dose of 75-mg showed maximal inhibition of mRNA expression of the four-CC chemokine receptor type-I dependent marker genes. chemokine ligand -2 and Peroxisome proliferator-activated receptor gamma-mRNAs by doses of 300 mg and higher, and for Ras-related protein rab-7b mRNA by doses of 500 mg and higher
Boehringer Ingelheim was developing BI-638683, a CCR1 antagonist, for the potential oral treatment of rheumatoid arthritis. A phase I trial was completed in December 2010 . Phase I data was presented in June 2014
Chemotactic Cytokine Receptor 1 (CCRl) belongs to a large family (>20) of chemotactic cytokine (chemokine) receptors that interact with specific chemokines (>50) to mediate leukocyte trafficking, granule exocytosis, gene transcription, mitogenic effects and apoptosis. Chemokines are best known for their ability to mediate basal and inflammatory leukocyte trafficking. The binding of at least three chemokines (MIP-1 alpha/CCL3, MCP3/CCL7 and RANTES/CCL5) to CCRl is responsible for the trafficking of monocytes, macrophages and THl cells to inflamed tissues of rheumatoid arthritis (RA) and multiple sclerosis (MS) patients (Trebst et al. (2001) American J of Pathology 159 p. 1701). Macrophage inflammatory protein 1 alpha (MIP-1 alpha), macrophage chemoattractant protein 3 (MCP-3) and regulated on activation, normal T-cell expressed and secreted (RANTES) are all found in the CNS of MS patients, while MIP-1 alpha and RANTES are found in the CNS in the experimental autoimmune encephalomyelitis (EAE) model of MS (Review: Gerard
and Rollins (2001) Nature Immunology). Macrophages and Thl cells in the inflamed synovia of RA patients are major producers of MIP-1 alpha and RANTES, which continuously recruit leukocytes to the synovial tissues of RA patients to propagate chronic inflammation (Volin et al. (1998) Clin. Immunol. Immunopathology; Koch et al. (1994) J. Clin. Investigation; Conlon et al. (1995) Eur. J. Immunology). Antagonizing the interactions between CCR1 and its chemokine ligands is hypothesized to block chemotaxis of monocytes, macrophages and Thl cells to inflamed tissues and thereby ameliorate the chronic inflammation associated with autoimmune diseases such as RA and MS.
Evidence for the role of CCR1 in the development and progression of chronic inflammation associated with experimental autoimmune encephalitis (EAE), a model of multiple sclerosis, is based on both genetic deletion and small molecule antagonists of CCR1. CCR1 deficient mice were shown to exhibit reduced susceptibility (55% vs. 100%) and reduced severity (1.2 vs. 2.5) of active EAE (Rottman et al. (2000) Eur. J. Immunology). Furthermore, administration of small molecule antagonist of CCR1, with moderate affinity (K; = 120 nM) for rat CCR1, was shown to delay the onset and reduce the severity of EAE when administered intravenously (Liang et al. (2000) /. Biol. Chemistry). Treatment of mice with antibodies specific for the CCR1 ligand MIP- 1 alpha have also been shown to be effective in preventing development of acute and relapsing EAE by reducing the numbers of T cells and macrophages recruited to the CNS (Karpus et al. (1995) /. Immunology; Karpus and Kennedy (1997) /. Leukocyte Biology). Thus, at least one CCR1 ligand has been demonstrated to recruit leukocytes to the CNS and propagate chronic inflammation in EAE, providing further in vivo validation for the role of CCR1 in EAE and MS.
In vivo validation of CCR1 in the development and propagation of chronic inflammation associated with RA is also significant. For example, administration of a CCR1 antagonist in the collagen induced arthritis model (CIA) in DBA/1 mice has been shown to be effective in reducing synovial inflammation and joint destruction (Plater-Zyberk et al. (1997) Immunology Letters). Another publication described potent antagonists of murine CCR1 that reduced severity (58%) in LPS-accelerated collagen-induced arthritis (CIA), when administered orally {Biorganic and Medicinal Chemistry Letters 15, 2005, 5160-5164). Published results from a Phase lb clinical trial with an oral CCRl antagonist demonstrated a trend toward clinical improvement in the absence of adverse side effects (Haringman et al. (2003) Ann. Rheum. Dis.). One third of the patients achieved a 20% improvement in rheumatoid arthritis signs and symptoms (ACR20) on day 18 and CCRl positive cells were reduced by 70% in the synovia of the treated patients, with significant reduction in specific cell types including 50% reduction in CD4+ T cells, 50% reduction in CD8+ T cells and 34% reduction in macrophages.
Studies such as those cited above support a role for CCRl in MS and RA and provide a therapeutic rationale for the development of CCRl antagonists.
Flow chemistry is also known as continuous flow or plug flow chemistry. It involves a chemical reaction run in a continuous flow stream. The process offers potential for the efficient manufacture of chemical products. Recent breakthroughs using Vapourtec systems are in production of Tamoxifen (Breast Cancer) and Artemisinin (Malaria).
Reactants are first pumped into a mixing device. Flow continues through a temperature controlled reactor until the reaction is complete. The reactor can be a simple pipe, tube or complex micro-structured device. The mixing device and reactor are maintained at the temperature to promote the desired reaction. The reactants may also be exposed to an electrical flux or a photon flux to promote an electrochemical or photochemical reaction.
The [3 + 2] dipolar cycloaddition of unstabilized azomethine ylides can be used to construct susbstituted pyrrolidines, which can be useful building blocks in pharmaceutical and natural product synthesis. A common experimental procedure involves in situ generation of the intermediate ylide in the presence of the dipolarophile; however, this technique can produce a significant exotherm when reactive dipolarophiles (e.g., acrylates, maleimides) are employed. Now Fray and co-workers at Pfizer describe efforts to conduct this chemistry using continuous flow technology, which is often beneficial when applied to systems involving highly energetic intermediates ( Tetrahedron Lett.2010, 51, 1026−1029).
The authors provide a description and schematic for the flow apparatus used and optimized the system for flow-rate/residence time, temperature, and pressure. It was found that the residence times could be reduced to 15 min if a higher operating temperature (100 °C) was used. The best conditions were then applied to a series of substrates, but it was determined that differences in reactivity required adjustment of parameters to achieve optimal results. For comparison, the authors also conducted the reaction in typical batch mode and found the yield to be higher. Nonetheless, the flow process was demonstrated to be capable of processing 30 g substrate within 1 h of operation (87% isolated yield after chromatography).
To demonstrate the viability of performing the cycloaddition in flow on a reasonable scale, the Vapourtec™ R2+/R4 was equipped with four heated reaction loops (total volume 40 ml) so that we could react compound 1 with ethyl acrylate under the previously optimised conditions (0.5 M overall in MeCN, 70 C, 10 min). From a reaction on 30 g scale, we obtained compound 3a in 87% yield, after chromatography in only 1 h.
(e) Srihari, P.; Yaragorla, S. R.; Basu, D.; Chandrasekhar, S. Synthesis 2006, 2646. 12. An attractive alternative has been described for generating the azomethine ylide via decarboxylation, for example, N-benzylglycine, paraformaldehyde, toluene, reflux, see: Joucla, M.; Mortier, J. Bull. Soc. Chim. Fr 1988, 579; Rodriguez Sarmiento, R. M.; Wirz, B.; Iding, H. Tetrahedron: Asymmetry 2003, 14, 1547.
Pfizer Global Research and Development, Sandwich, Kent CT13 9NJ, United Kingdom
Abstract
The [3+2] dipolar cycloaddition reactions of the unstabilised azomethine ylide precursor benzyl(methoxymethyl)(trimethylsilylmethyl)amine with 12 electron-deficient alkenes in the presence of catalytic trifluoroacetic acid are examined under continuous flow conditions (20–100 °C, 10–60 min residence time). The more reactive and hazardous alkenes such as ethyl acrylate, N-methylmaleimide and (E)-2-nitrostyrene afford substituted N-benzylpyrrolidine products in 77–83% yields, whereas less reactive dipolarophiles such as (E)-crotononitrile and ethyl methacrylate give lower yields (59–63%). Under optimised conditions, the reaction with ethyl acrylate is scaled up to afford ethyl N-benzylpyrrolidine-3-carboxylate (30 g, 87%) in 1 h.
Under continuous flow conditions an azomethine ylide precursor reacts with 12 electron-deficient alkenes to give the corresponding pyrrolidines. The most reactive and therefore hazardous dipolarophiles give the best yields.
MORE INSIGHT………..
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen.[1]Hence, the reaction is sometimes referred to as the Huisgen cycloaddition (this term is often used to specifically describe the 1,3-dipolar cycloaddition between an organic azide and an alkyne to generate 1,2,3-triazole). Currently, 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives.
Mechanistic overview
There were originally two proposals that describe the mechanism of the 1,3-dipolar cycloaddition: first, the concertedpericycliccycloaddition mechanism, proposed by Rolf Huisgen;[2] and second, the stepwise mechanism involving a diradicalintermediate, proposed by Firestone.[3] After much debate, the former proposal is now generally accepted[4]—the 1,3-dipole reacts with the dipolarophile in a concerted, often asynchronous, and symmetry-allowed π4s + π2s fashion through a thermal six-electron Huckel aromatic transition state. Although, there are few examples of stepwise mechanism of the catalyst free 1,3-dipolar cycloaddition reactions for thiocarbonyl ylides,[5] and nitrile oxides[6]
Pericyclic mechanism
Huisgen investigated a series of cycloadditions between the 1,3-dipolar diazo compounds and various dipolarophilic alkenes.[2] The following observations support the concerted pericyclic mechanism, and refute the stepwise diradical or the stepwise polar pathway.
Substituent effects: Different substituents on the dipole do not exhibit a large effect on the cycloaddition rate, suggesting that the reaction does not involve a charge-separated intermediate.
Solvent effects: Solvent polarity has little effect on the cycloaddition rate, in line with the pericyclic mechanism where polarity does not change much in going from the reactants to the transition state.
Stereochemistry: 1,3-dipolar cycloadditions are always stereospecific with respect to the dipolarophile (i.e., cis-alkenes giving syn-products), supporting the concerted pericyclic mechanism in which two sigma bonds are formed simultaneously.
Structure and nomenclature of all second-row 1,3-dipoles consisting of carbon, nitrogen and oxygen centers. The dipoles are categorized as allyl-type or propargyl/allenyl-type based on the geometry of the central atom.
A 1,3-dipole is an organic molecule that can be represented as either an allyl-type or a propargyl/allenyl-type zwitterionic octet/sextet structures. Both types of 1,3-dipoles share four electrons in the π-system over three atoms. The allyl-type is bent whereas the propargyl/allenyl-type is linear in geometry. There are a total of 18 second-row 1,3-dipoles (see structures in the thumbnail on the right).[7]1,3-Dipoles containing higher-row elements such as sulfur or phosphorus are also known, but are utilized less routinely.
Resonance structures can be drawn to delocalize both negative and positive charges onto any terminus of a 1,3-dipole (see the scheme below). A more accurate method to describe the electronic distribution on a 1,3-dipole is to assign the major resonance contributor based on experimental or theoretical data, such as dipole moment measurements[8] or computations.[9] For example, diazomethane bears the largest negative character at the terminal nitrogen atom, while hydrazoic acid bears the largest negative character at the internal nitrogen atom.
Consequently, this ambivalence means that the termini of a 1,3-dipole can be treated as both nucleophilic and electrophilic at the same time. The extent of nucleophilicity and electrophilicity at each terminus can be evaluated using the frontier molecular orbitals, which can be obtained computationally. In general, the atom that carries the largest orbital coefficient in the HOMO acts as the nucleophile, whereas that in the LUMO acts as the electrophile. The most nucleophilic atom is usually, but not always, the most electron-rich atom.[10][11][12] In 1,3-dipolar cycloadditions, identity of the dipole-dipolarophile pair determines whether the HOMO or the LUMO character of the 1,3-dipole will dominate (see discussion on frontier molecular orbitals below).
Dipolarophile
The most commonly used dipolarophiles are alkenes and alkynes. Heteroatom-containing dipolarophiles such as carbonyls and imines can also undergo 1,3-dipolar cycloaddition. Other examples of dipolarophiles include fullerenes and nanotubes, which can undergo 1,3-dipolar cycloaddition with azomethine ylide in the Prato reaction.
Solvent effects
1,3-dipolar cycloadditions experience very little solvent effect because both the reactants and the transition states are generally non-polar. For example, the rate of reaction between phenyl diazomethane and ethyl acrylate or norbornene (see scheme below) changes only slightly upon varying solvents from cyclohexane to methanol.[13]
Lack of solvent effects in 1,3-dipolar cycloaddition is clearly demonstrated in the reaction between enamines and dimethyl diazomalonate (see scheme below).[14] The polar reaction, N-cyclopentenyl pyrrolidine nucleophilic addition to the diazo compound, proceeds 1,500 times faster in polar DMSO than in non-polar decalin. On the other hand, a close analog of this reaction, N-cyclohexenyl pyrrolidine 1,3-dipolar cycloaddition to dimethyl diazomalonate, is sped up only 41-fold in DMSO relative to decalin.
Frontier molecular orbital theory
1,3-Dipolar cycloadditions are pericyclic reactions, which obey the Dewar-Zimmerman rules and the Woodward–Hoffmann rules. In the Dewar-Zimmerman treatment, the reaction proceeds through a 5-center, zero-node, 6-electron Huckel transition state for this particular molecular orbital diagram. However, each orbital can be randomly assigned a sign to arrive at the same result. In the Woodward–Hoffmann treatment, frontier molecular orbitals (FMO) of the 1,3-dipole and the dipolarophile overlap in the symmetry-allowed π4s + π2s manner. Such orbital overlap can be achieved in three ways: type I, II and III.[15] The dominant pathway is the one which possesses the smallest HOMO-LUMO energy gap.
Type I
The dipole has a high-lying HOMO which overlaps with LUMO of the dipolarophile. A dipole of this class is referred to as a HOMO-controlled dipole or a nucleophilic dipole, which includes azomethine ylide, carbonyl ylide, nitrile ylide, azomethine imine, carbonyl imine and diazoalkane. These dipoles add to electrophilic alkenes readily. Electron-withdrawing groups (EWG) on the dipolarophile would accelerate the reaction by lowering the LUMO, while electron-donating groups (EDG) would decelerate the reaction by raising the HOMO. For example, the reactivity scale of diazomethane against a series of dipolarophiles is shown in the scheme below. Diazomethane reacts with the electron-poor ethyl acrylate more than a million times faster than the electron rich butyl vinyl ether.[16]
This type resembles the normal-electron-demand Diels-Alder reaction, in which the diene HOMO combines with the dienophile LUMO.
Type II
HOMO of the dipole can pair with LUMO of the dipolarophile; alternatively, HOMO of the dipolarophile can pair with LUMO of the dipole. This two-way interaction arises because the energy gap in either direction is similar. A dipole of this class is referred to as a HOMO-LUMO-controlled dipole or an ambiphilic dipole, which includes nitrile imide, nitrone, carbonyl oxide, nitrile oxide, and azide. Any substituent on the dipolarophile would accelerate the reaction by lowering the energy gap between the two interacting orbitals; i.e., an EWG would lower the LUMO while an EDG would raise the HOMO. For example, azides react with various electron-rich and electron-poor dipolarophile with similar reactivities (see reactivity scale below).[17]
Type III
The dipole has a low-lying LUMO which overlaps with HOMO of the dipolarophile (indicated by red dashed lines in the diagram). A dipole of this class is referred to as a LUMO-controlled dipole or an electrophilic dipole, which includes nitrous oxide and ozone. EWGs on the dipolarophile decelerate the reaction, while EDGs accelerate the reaction. For example, ozone reacts with the electron-rich 2-methylpropene about 100,000 times faster than the electron-poor tetrachloroethene (see reactivity scale below).[18]
Concerted processes such as the 1,3-cycloaddition require a highly ordered transition state (high negative entropy of activation) and only moderate enthalpy requirements. Using competition reaction experiments, relative rates of addition for different cycloaddition reactions have been found to offer general findings on factors in reactivity.
Conjugation, especially with aromatic groups, increases the rate of reaction by stabilization of the transition state. During the transition, the two sigma bonds are being formed at different rates, which may generate partial charges in the transition state that can be stabilized by charge distribution into conjugated substituents.
More polarizable dipolarophiles are more reactive because diffuse electron clouds are better suited to initiate the flow of electrons.
Dipolarophiles with high angular strain are more reactive due to increased energy of the ground state.
Increased steric hindrance in the transition state as a result of unhindered reactants dramatically lowers the reaction rate.
Hetero-dipolarophiles add more slowly, if at all, compared to C,C-diapolarophiles due to a lower gain in sigma bond energy to offset the loss of a pi bond during the transition state.
Isomerism of the dipolarophile affects reaction rate due to sterics. trans-isomers are more reactive (trans-stilbene will add diphenyl(nitrile imide) 27 times faster than cis-stilbene) because during the reaction, the 120° bond angle shrinks to 109°, bringing eclipsing cis-substituents towards each other for increased steric clash.
Stereospecificity
1,3-dipolar cycloadditions usually result in retention of configuration with respect to both the 1,3-dipole and the dipolarophile. Such high degree of stereospecificity is a strong support for the concerted over the stepwise reaction mechanisms. As mentioned before, there are many examples that show that the reactions were stepwise, thus, presenting partial or no stereospecificity.
With respect to dipolarophile
cis-Substituents on the dipolarophilic alkene end up cis, and trans-substituents end up trans in the resulting five-membered cyclic compound (see scheme below).[19]
With respect to dipole
Generally, the stereochemistry of the dipole is not of major concern because only few dipoles could form stereogenic centers, and resonance structures allow bond rotation which scrambles the stereochemistry. However, the study of azomethine ylides has verified that cycloaddition is also stereospecific with respect to the dipole component. Diastereopure azomethine ylides are generated via electrocyclic ring opening of aziridines, and then rapidly trapped with strong dipolarophiles before bond rotation can take place (see scheme below).[20][21] If weaker dipolarophiles are used, bonds in the dipole have the chance to rotate, resulting in impaired cycloaddition stereospecificity.
These results altogether confirm that 1,3-dipolar cycloaddition is stereospecific, giving retention of both the 1,3-dipole and the dipolarophile.
Diastereoselectivity
When two or more chiral centers are generated during the reaction, diastereomeric transition states and products can be obtained. In the Diels-Alder cycloaddition, the endodiastereoselectivity due to secondary orbital interactions is usually observed. In 1,3-dipolar cycloadditions, however, there are two forces that influence the diastereoselectivity: the attractive π-interaction (resembling secondary orbital interactions in the Diels-Alder cycloaddition) and the repulsive steric interaction. Unfortunately, these two forces often cancel each other, causing poor diastereoselection in 1,3-dipolar cycloaddition.
Examples of substrate-controlled diastereoselective 1,3-dipolar cycloadditions are shown below. First is the reaction between benzonitrile N-benzylide and methyl acrylate. In the transition state, the phenyl and the methyl ester groups stack to give the cis-substitution as the exclusive final pyrroline product. This favorable π-interaction offsets the steric repulsion between the phenyl and the methyl ester groups.[22] Second is the reaction between nitrone and dihydrofuran. The exo-selectivity is achieved to minimize steric repulsion.[23] Last is the intramolecular azomethine ylide reaction with alkene. The diastereoselectivity is controlled by the formation of a less strained cis–fused ring system.[24]
Directed 1,3-dipolar cycloaddition
Trajectory of the cycloaddition can be controlled to achieve a diastereoselective reaction. For example, metals can chelate to the dipolarophile and the incoming dipole and direct the cycloaddition selectively on one face. The example below shows addition of nitrile oxide to an enantiomerically pure allyl alcohol in the presence of a magnesium ion. The most stable conformation of the alkene places the hydroxyl group above the plane of the alkene. The magnesium then chelates to the hydroxyl group and the oxygen atom of nitrile oxide. The cycloaddition thus comes from the top face selectively.[25]
Such diastereodirection has been applied in the synthesis of epothilones.[26]
The dominant electronic interaction is the combination between the largest HOMO orbital and the largest LUMO orbital. Therefore, regioselectivity is governed by the atoms that bear the largest orbital HOMO and LUMO coefficients.[28][29]
For example, consider the cycloaddition of diazomethane to three dipolarophiles: methyl acrylate, styrene or methyl cinnamate. The carbon of diazomethane bears the largest HOMO orbital, while the terminal olefinic carbons of methyl acrylate and styrene bear the largest LUMO orbital. Hence, cycloaddition gives the substitution at the C-3 position regioselectively. For methyl cinnamate, the two substituents (Ph v.s. COOMe) compete at withdrawing electrons from the alkene. The carboxyl is the better electron-withdrawing group, causing the β-carbon to be most electrophilic. Thus, cycloaddition yields the carboxyl group on C-3 and the phenyl group on C-4 regioselectively.
Steric effect
Steric effects can either cooperate or compete with the aforementioned electronic effects. Sometimes steric effects completely outweighs the electronic preference, giving the opposite regioisomer exclusively.[30]
For example, diazomethane generally adds to methyl acrylate to give 3-carboxyl pyrazoline. However, by putting more steric demands into the system, we start to observe the isomeric 4-carboxyl pyrazolines. The ratio of these two regioisomers depends on the steric demands. At the extreme, increasing the size from hydrogen to t-butyl shifts the regioselectivity from 100% 3-carboxyl to 100% 4-carboxyl substitution.[31][32]
Synthetic Applications
1,3-dipolar cycloadditions are important routes toward the synthesis of many important 5-membered heterocycles such as triazoles, furans, isoxazoles, pyrrolidines, and others. Additionally, some cycloadducts can be cleaved to reveal the linear skeleton, providing another route toward the synthesis of aliphatic compounds. These reactions are tremendously useful also because they are stereospecific, diastereoselective and regioselective. Several examples are provided below.
Nitrile oxides
1,3-dipolar cycloaddition with nitrile oxides is a widely used masked-aldol reaction. Cycloaddition between a nitrile oxide and an alkene yields the cyclic isoxazoline product, whereas the reaction with an alkyne yields the isoxazole. Both isoxazolines and isoxazoles can be cleaved by hydrogenation to reveal aldol-type β-hydroxycarbonyl or Claisen-type β-dicarbonyl products, respectively.
Nitrile oxide-alkyne cycloaddition followed by hydrogenation was utilized in the synthesis of Miyakolide as illustrated in the figure below.[33]
Carbonyl ylides
1,3-dipolar cycloaddition reactions have emerged as powerful tools in the synthesis of complex cyclic scaffolds and molecules for medicinal, biological, and mechanistic studies. Among them, [3+2] cycloaddition reactions involving carbonyl ylides have extensively been employed to generate oxygen-containing five-membered cyclic molecules.[34]
Preparation of Carbonyl Ylides for 1,3-Dipolar Cycloaddition Reactions
Ylides are regarded as positively charged heteroatoms connected to negatively charged carbon atoms, which include ylides of sulfonium, thiocarbonyl, oxonium, nitrogen, and carbonyl.[35] Several methods exist for generating carbonyl ylides, which are necessary intermediates for generating oxygen-containing five-membered ring structures, for [3+2] cycloaddition reactions.
Synthesis of Carbonyl Ylides from Diazomethane Derivatives by Photocatalysis
Another early example of carbonyl ylide synthesis by photocatalysis was reported by Olah et al.[37] Dideuteriodiazomethane was photolysed in the presence of formaldehyde to generate the dideuterioformaldehyde carbonyl ylide.
Synthesis of Carbonyl Ylides from Hydroxypyrones via Proton Transfer
Carbonyl ylides can be synthesized by acid catalysis of hydroxy-3-pyrones in the absence of a metal catalyst.[38] An initial tautomerization occurs, followed by elimination of the leaving group to aromatize the pyrone ring and to generate the carbonyl ylide. A cycloaddition reaction with a dipolarophile lastly forms the oxacycle. This approach is less widely employed due to its limited utility and requirement for pyrone skeletons.
5-hydroxy-4-pyrones can also be used to synthesize carbonyl ylides via an intramolecularhydrogen transfer.[39] After hydrogen transfer, the carbonyl ylide can then react with dipolarophiles to form oxygen-containing rings.
Synthesis of α-Halocarbonyl Ylides from Dihalocarbenes
Dihalocarbenes have also been employed to generate carbonyl ylides. The electron withdrawing nature of dihalocarbenes has been exploited by Landgrebe and coworkers for this purpose.[40][41][42] Both phenyl(bromodichloromethyl)mercury and phenyl(tribromomethyl)mercury have been converted to dichlorocarbenes and dibromocarbenes, respectively. The carbonyl ylide can be generated upon reaction of the dihalocarbenes with ketones or aldehydes. However, the synthesis of α-halocarbonyl ylides can also undesirably lead to the loss of carbon monoxide and the generation of the deoxygenation product.
Synthesis of Carbonyl Ylides from Diazomethane Derivatives by Metal Catalysis
A universal approach for generating carbonyl ylides involves metal catalysis of α-diazocarbonyl compounds, generally in the presence of dicopper or dirhodium catalysts.[43] After release of nitrogen gas and conversion to the metallocarbene, an intermolecular reaction with a carbonyl group can generate the carbonyl ylide. Subsequent cycloaddition reaction with an alkene or alkyne dipolarophile can afford oxygen-containing five-membered rings. Popular catalysts that give modest yields towards synthesizing oxacycles include Rh2(OAc)4 and Cu(acac)2.[44][45]
Mechanism of the 1,3-Dipolar Cycloaddition Reaction Mediated by Metal Catalysis of Diazocarbonyl Compounds
The universality and extensive use of 1,3-dipolar cycloaddition reactions mediated by metal catalysis of diazocarbonyl molecules, for synthesizing oxygen-containing five-membered rings, has spurred significant interest into its mechanism. Several groups have investigated the mechanism to expand the scope of synthetic molecules with respect to regio- and stereo-selectivity. However, due to the high turn over frequencies of these reactions, the intermediates and mechanism remains elusive. The generally accepted mechanism, developed by characterization of stable ruthenium-carbenoid complexes[46] and rhodium metallocarbenes,[47] involves an initial formation of a metal-carbenoid complex from the diazo compound. Elimination of nitrogen gas then affords a metallocarbene. An intramolecular nucleophilic attack by the carbonyl oxygen regenerates the metal catalyst and forms the carbonyl ylide. The carbonyl ylide can then react with an alkene or alkyne, such as dimethyl acetylenedicarboxylate (DMAD) to generate the oxacycle.
However, it is uncertain whether the metallocarbene intermediate generates the carbonyl ylide. In some cases, metallocarbenes can also react directly with dipolarophiles.[48] In these cases, the metallocarbene, such as the dirhodium(II)tetracarboxylate carbene, is stabilized through hyperconjugative metal enolate-type interactions.[49][50][51][52]Subsequent 1,3-dipolar cycloaddition reaction occurs through a transient metal-complexed carbonyl ylide. Therefore, a persistent metallocarbene can influence the stereoselectivity and regioselectivity of the 1,3-dipolar cycloaddition reaction based on the stereochemistry and size of the metal ligands.
Regioselectivity of the 1,3-Dipolar Cycloaddition Reaction Mediated by Metal Catalysis of Diazocarbonyl Compounds
Regioselectivity of 1,3-dipolar cycloaddition reactions between carbonyl ylide dipoles and alkynyl or alkenyl dipolarophiles is essential for generating molecules with defined regiochemistry. FMO theory and analysis of the HOMO-LUMO energy gaps between the dipole and dipolarophile can rationalize and predict the regioselectivity of experimental outcomes.[55][56] The HOMOs and LUMOs can belong to either the dipole or dipolarophile, for which HOMOdipole-LUMOdipolarophile or HOMOdipolarophile-LUMOdipole interactions can exist. Overlap of the orbitals with the largest coefficients can ultimately rationalize and predict results.
The archetypal regioselectivity of the 1,3-dipolar cycloaddition reaction mediated by carbonyl ylide dipoles has been examined by Padwa and coworkers.[54][57] Using a Rh2(OAc)4catalyst in benzene, diazodione underwent a 1,3-dipolar cycloaddition reaction with methyl propiolate and methyl propargylether. The reaction with methyl propiolate affords two regioisomers with the major resulting from the HOMOdipole-LUMOdipolarophile interaction, which has the largest coefficients on the carbon proximal to the carbonyl group of the carbonyl ylide and on the methyl propiolate terminal alkyne carbon. The reaction with methyl propargyl ether affords one regioisomer resulting from the HOMOdipolarophile-LUMOdipole interaction, which has largest coefficients on the carbon distal to the carbonyl group of the carbonyl ylide and on the methyl propargyl ether terminal alkyne carbon.
Regioselectivities of 1,3-dipolar cycloaddition reactions mediated by metal catalysis of diazocarbonyl compounds may also be influenced by the metal through formation of stable metallocarbenes.[48][58] Stabilization of the metallocarbene, via metal enolate-type interactions, will prevent the formation of carbonyl ylides, resulting in a direct reaction between the metallocarbene dipole and an alkynyl or alkenyl dipolarophile (see image of The dirhodium(II)tetracarboxylate metallocarbene stabilized by πC-Rh→πC=O hyperconjugation.). In this situation, the metal ligands will influence the regioselectivity and stereoselectivity of the 1,3-dipolar cycloaddition reaction.
Stereoselectivity and Asymmetric Induction of the 1,3-Dipolar Cycloaddition Reaction Mediated by Metal Catalysis of Diazocarbonyl Compounds
The stereoselectivity of 1,3-dipolar cycloaddition reactions between carbonyl ylide dipoles and alkenyl dipolarophiles has also been closely examined. For alkynyl dipolarophiles, stereoselectivity is not an issue as relatively planar sp2 carbons are formed, while regioselectivity must be considered (see image of the Products of the 1,3-Dipolar Cycloaddition Reaction Between Carbonyl Ylide Dipoles and Alkenyl or Alkynyl Dipolarophiles). However, for alkenyl dipolarophiles, both regioselectivity and stereoselectivity must be considered as sp3 carbons are generated in the product species.
1,3-dipolar cycloaddition reactions between carbonyl ylide dipoles and alkenyl dipolarophiles can generate diastereomeric products.[52] The exo product is characterized with dipolarophile substituents being cis to the ether bridge of the oxacycle. The endo product is characterized with the dipolarophile substituents being trans to the ether bridge of the oxacycle. Both products can be generated through pericyclic transitions states involving concerted synchronous or concerted asynchronous processes.
One early example conferred stereoselectivity in terms of endo and exo products with metal catalysts and Lewis acids.[59] Reactions with just the metal catalyst Rh2(OAc)4 prefer the exo product while reactions with the additional Lewis acid Yb(OTf)3 prefer the endo product. The endo selectivity observed for Lewis acid cycloaddition reactions is attributed to the optimized orbital overlap of the carbonyl π systems between the dipolarophile coordinated by Yb(Otf)3 (LUMO) and the dipole (HOMO). After many investigations, two primary approaches for influencing the stereoselectivity of carbonyl ylide cycloadditions have been developed that exploit the chirality of metal catalysts and Lewis acids.[52]
The first approach employs chiral metal catalysts to modulate the endo and exo stereoselectivity. The chiral catalysts, in particular Rh2[(S)-DOSP]4 and Rh2[(S)-BPTV]4 can induce modest asymmetric induction and was used to synthesize the antifungal agent pseudolaric acid A.[60] This is a result of the chiral metal catalyst remaining associated with the carbonyl ylide during the cycloaddition, which confers facial selectivity. However, the exact mechanisms are not yet fully understood.
The second approach employs a chiral Lewis acid catalyst to induce facial stereoselectivity after the generation of the carbonyl ylide using an achiral metal catalyst.[61] The chiral Lewis acid catalyst is believed to coordinate to the dipolarophile, which lowers the LUMO of the dipolarophile while also leading to enantioselectivity.
1,3-Dipolar cycloaddition between an azomethine ylide and an alkene furnishes an azacyclic structure, such as pyrrolidine. This strategy has been applied to the synthesis of spirotryprostatin A.[62]
The 1,3-dipolar cycloaddition between organic azides and terminal alkynes (i.e., the Huisgen cycloaddition) has been widely utilized for bioconjugation.
Copper catalysis
The Huisgen reaction generally does not proceed readily under mild conditions. Meldal et al. and Sharpless et al. independently developed a copper(I)-catalyzed version of the Huisgen reaction, CuAAC (for Copper-catalyzed Azide-Alkyne Cycloaddition), which proceeds very readily in mild, including physiological, conditions (neutral pH, room temperature and aqueous solution).[63][64] This reaction is also bioorthogonal: azides and alkynes are both generally absent from biological systems and therefore these functionalities can be chemoselectively reacted even in the cellular context. They also do not react with other functional groups found in Nature, so they do not perturb biological systems. The reaction is so versatile that it is termed the “Click” chemistry. Although copper(I) is toxic, many protective ligands have been developed to both reduce cytotoxicity and improve rate of CuAAC, allowing it to be used in in vivo studies.[65]
For example, Bertozzi et al. reported the metabolic incorporation of azide-functionalized sugars into the glycan of the cell membrane, and subsequent labeling with fluorophore-alkyne conjugate. The now fluorescently labeled cell membrane can be imaged under the microscope.[66]
Strain-promoted cycloaddition
To avoid toxicity of copper(I), Bertozzi et al. developed the Strain-Promoted Azide-Alkyne Cycloaddition (SPAAC) between organic azide and strained cyclooctyne. The angle distortion of the cyclooctyne helps to speed up the reaction, enabling it to be used in physiological conditions without the need for the catalyst.[67]
For instance, Ting et al. introduced an azido functionality onto specific proteins on the cell surface using a ligase enzyme. The azide-tagged protein is then labeled with cyclooctyne-fluorophore conjugate to yield a fluorescently labeled protein.[68]
Department of Chemistry, Kwame Nkrumah University of Science and Technology, Kumasi, Ghana
*Corresponding Author:
Richard Tia
Department of Chemistry
Kwame Nkrumah University of Science and Technology
Kumasi, Ghana Tel: 00233(0) 243574146 E-mail: richtiagh@yahoo.com
Received date: May 12, 2014; Accepted date: Septem
Jump up^Padwa, Albert (1983). 1,3-Dipolar Cycloaddition Chemistry. General Heterocyclic Chemistry Series. 1. United States of America: Wiley-Interscience. pp. 141–145. ISBN0-471-08364-X.
Jump up^Koszinowski, J. (1980). thesis (Ph.D. Thesis).
Jump up^Synthetic Reactions of M=C and M=N Bonds: Ylide Formation, Rearrangement, and 1,3-Dipolar Cycloaddition; Hiyama, T. W., J., Ed.; Elsevier, 2007; Vol. 11.
Jump up^Agard, Nicholas; Prescher, Jennifer; Bertozzi, Carolyn (2004). “A Strain-Promoted [3 + 2] Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems”. Journal of the American Chemical Society. 46: 15046–15047. doi:10.1021/ja044996f. PMID15547999.















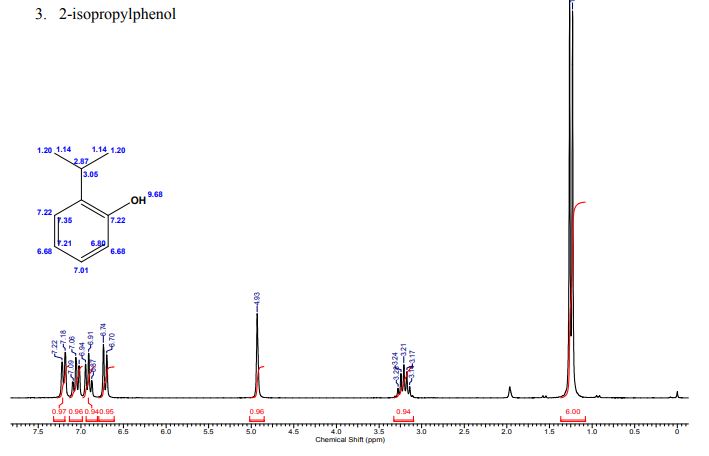
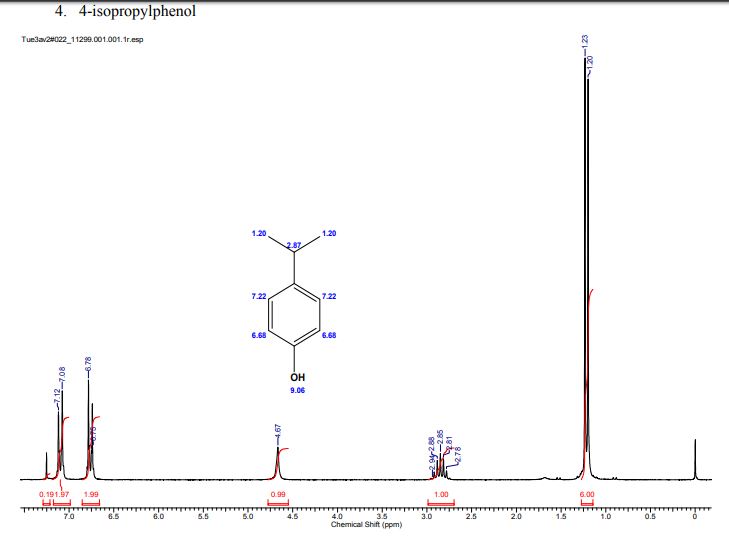



 READ
READ
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com




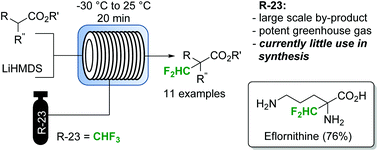
 Open Access
Open Access
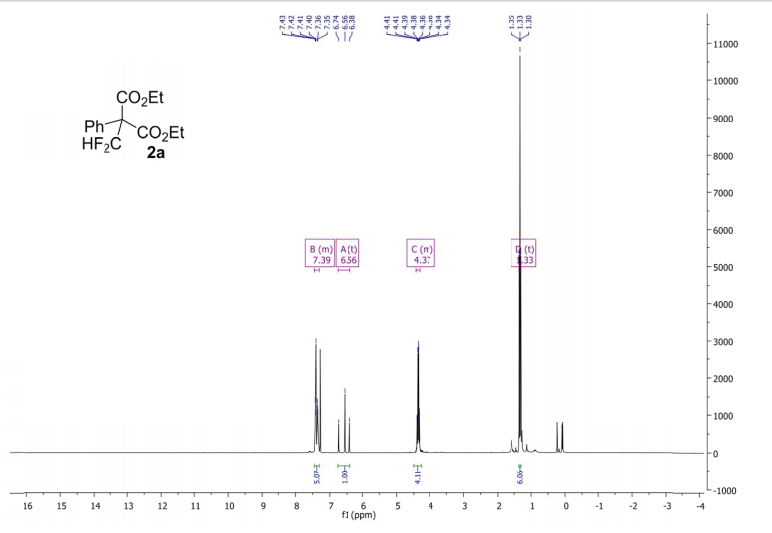
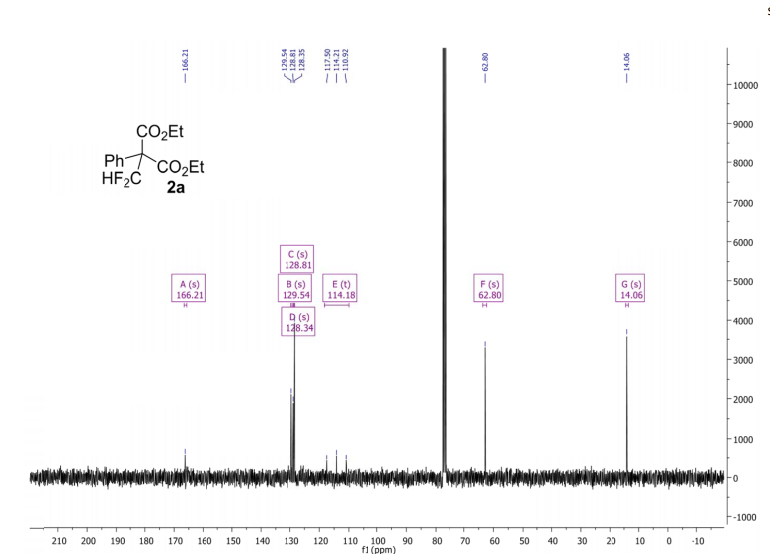
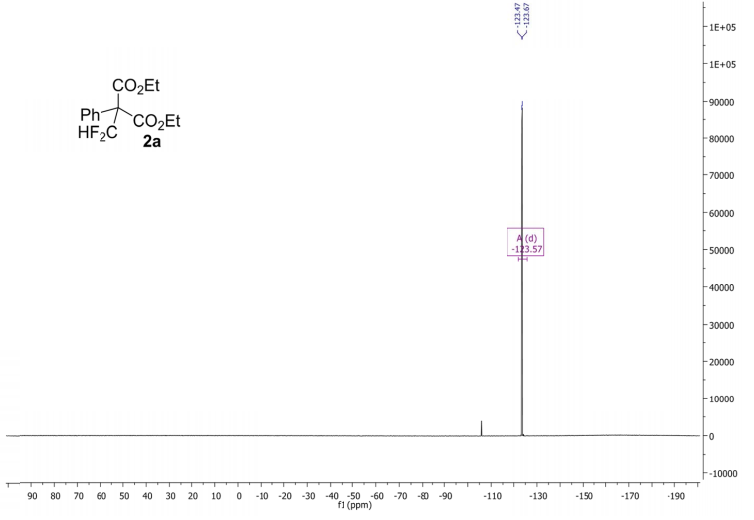


![[1860-5397-13-51-1]](/bjoc/content/figures/1860-5397-13-51-1.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i1]](/bjoc/content/inline/1860-5397-13-51-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i2]](/bjoc/content/inline/1860-5397-13-51-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i3]](/bjoc/content/inline/1860-5397-13-51-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i4]](/bjoc/content/inline/1860-5397-13-51-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i5]](/bjoc/content/inline/1860-5397-13-51-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i6]](/bjoc/content/inline/1860-5397-13-51-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i7]](/bjoc/content/inline/1860-5397-13-51-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-2]](/bjoc/content/figures/1860-5397-13-51-2.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i8]](/bjoc/content/inline/1860-5397-13-51-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i9]](/bjoc/content/inline/1860-5397-13-51-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i10]](/bjoc/content/inline/1860-5397-13-51-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i11]](/bjoc/content/inline/1860-5397-13-51-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i12]](/bjoc/content/inline/1860-5397-13-51-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i13]](/bjoc/content/inline/1860-5397-13-51-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i14]](/bjoc/content/inline/1860-5397-13-51-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i15]](/bjoc/content/inline/1860-5397-13-51-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i16]](/bjoc/content/inline/1860-5397-13-51-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i17]](/bjoc/content/inline/1860-5397-13-51-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i18]](/bjoc/content/inline/1860-5397-13-51-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i19]](/bjoc/content/inline/1860-5397-13-51-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i20]](/bjoc/content/inline/1860-5397-13-51-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i21]](/bjoc/content/inline/1860-5397-13-51-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i22]](/bjoc/content/inline/1860-5397-13-51-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-i23]](/bjoc/content/inline/1860-5397-13-51-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-3]](/bjoc/content/figures/1860-5397-13-51-3.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-13-51-4]](/bjoc/content/figures/1860-5397-13-51-4.jpg?scale=2.0&max-width=1024&background=FFFFFF)
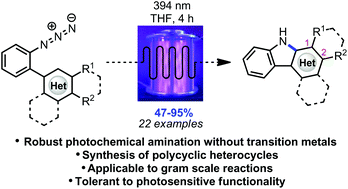

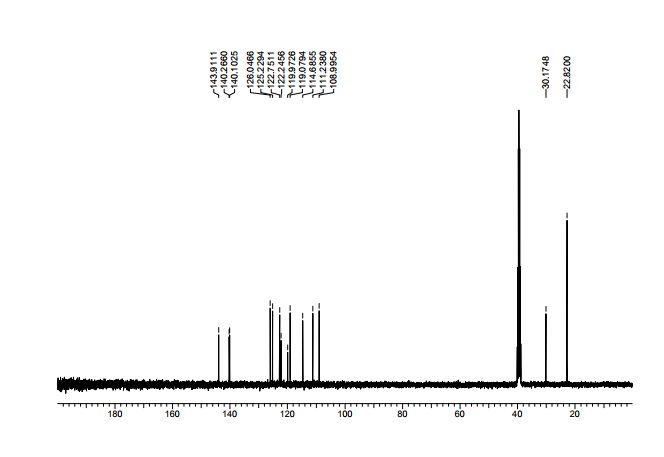
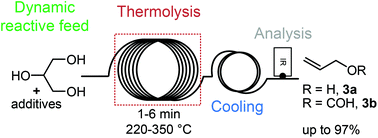



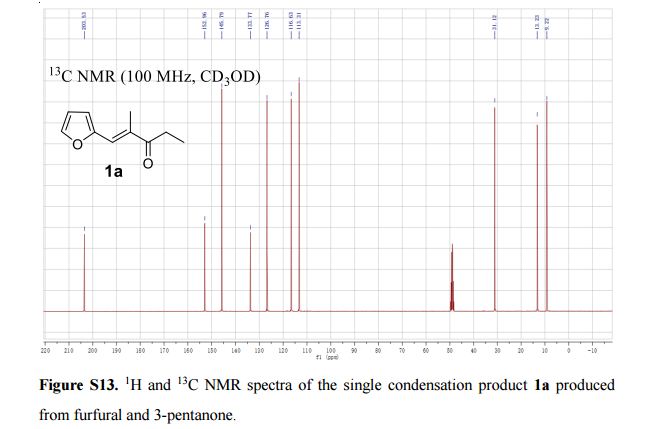
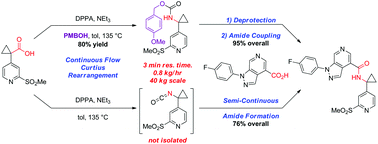
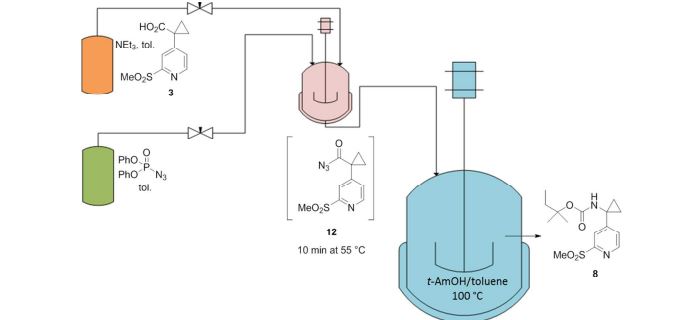
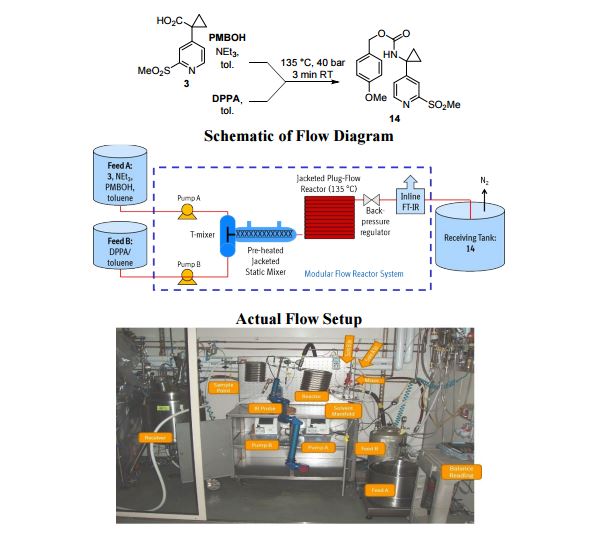

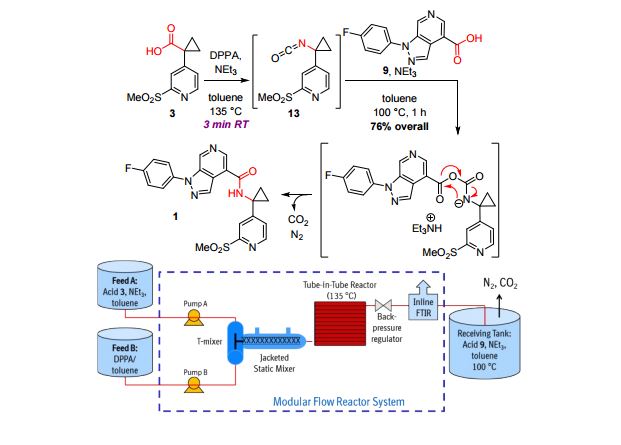
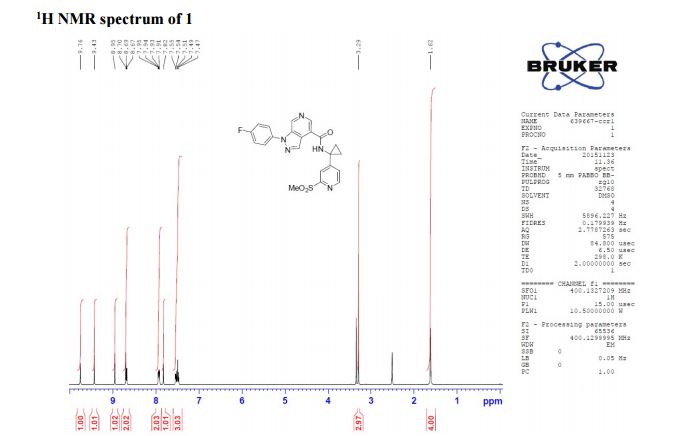
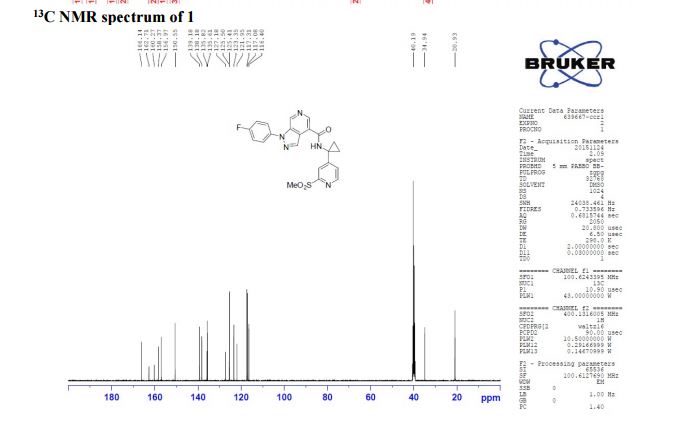
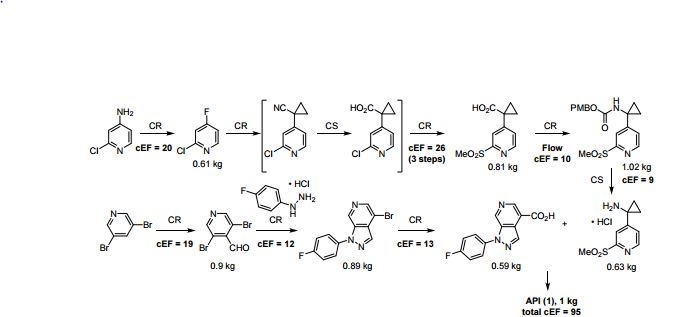
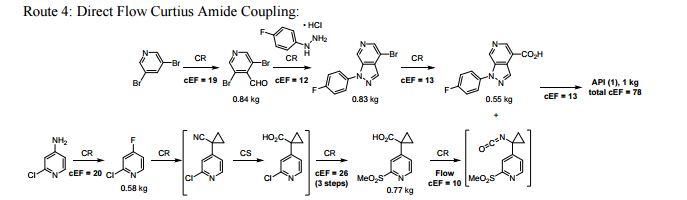
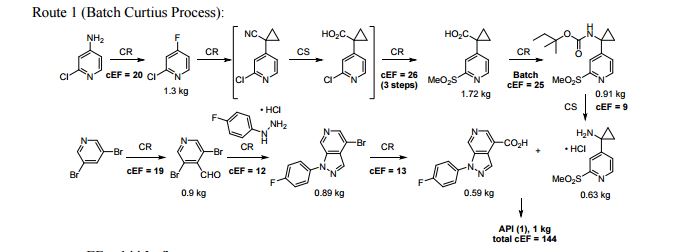
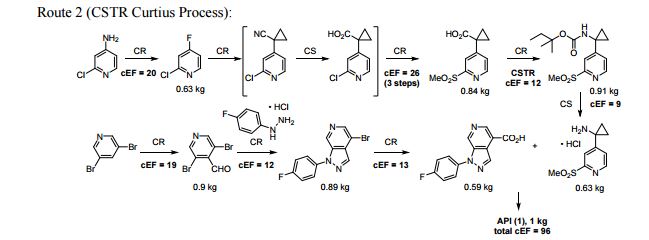
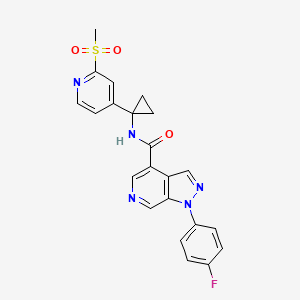
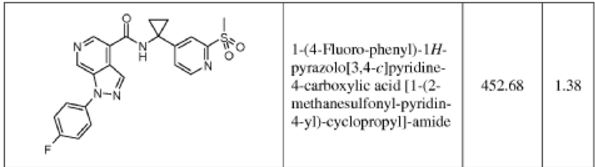

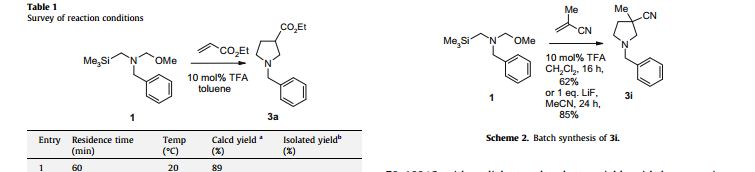







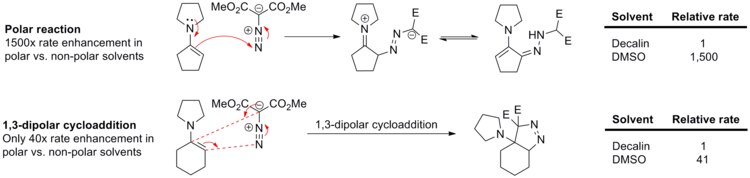
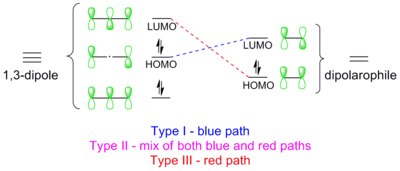
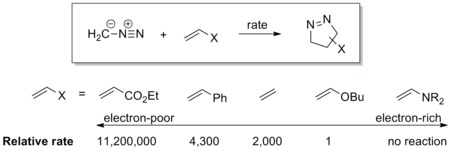




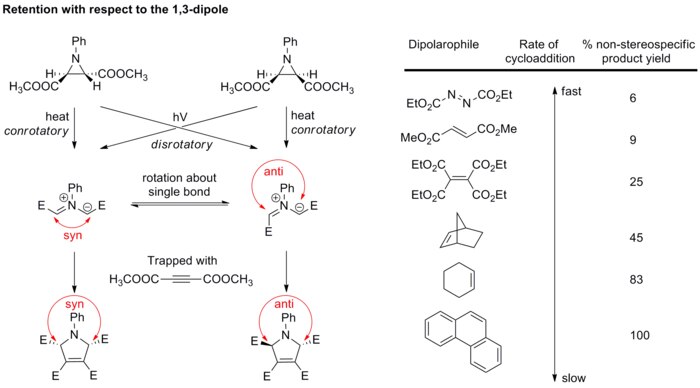

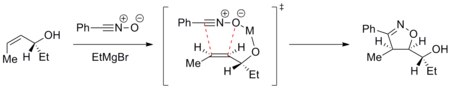











tetracarboxylate_metallocarbene_stabilized_by_%CF%80C-Rh%E2%86%92%CF%80C%3DO_hyperconjugation..png)







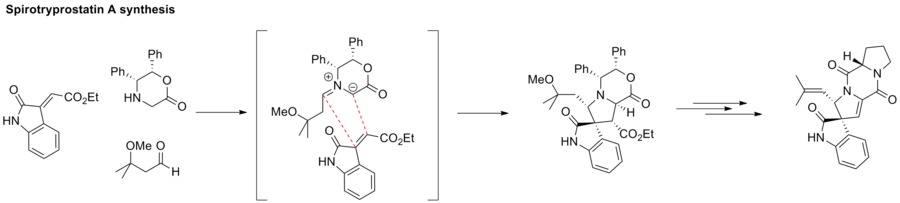




![Image result for [3 + 2] Dipolar Cycloadditions](https://www.omicsonline.org/articles-images/theoretical-computational-science-Dipolar-Cycloaddition-Reaction-2-117-s002.png)