











 A novel nanoparticle-based drug delivery system prevents the premature release of therapeutic compounds
A novel nanoparticle-based drug delivery system prevents the premature release of therapeutic compounds
Drug Release: Double the Control
drugs
Comments Off on Drug Release: Double the Control
Sep 202013

The bark of Nauclea latifolia contains tramadol at medicinal concentrations © imagebroker / Alamy
http://www.rsc.org/chemistryworld/2013/09/african-plant-natural-source-tramadol
In another example of nature beating chemists, the African plant Nauclea latifolia has been found to be a natural source of the synthetic opioid tramadol. First marketed in 1977, tramadol is frequently used to relive moderate to moderately-severe pain. While other synthetic drugs have later been found in nature, this is the first instance where the discovery involves clinically viable concentrations.
Colloquially known as the ‘African peach’ or ‘pin cushion tree’, N. latifolia is a flowering, sub-Saharan evergreen that grows widely across Central and West Africa and is used by local populations to treat a wide variety of ailments – including epilepsy, malaria, general pain and many infectious diseases………………………. READ ALL AT
http://www.rsc.org/chemistryworld/2013/09/african-plant-natural-source-tramadol

tramadol

tramadol hydrocloride
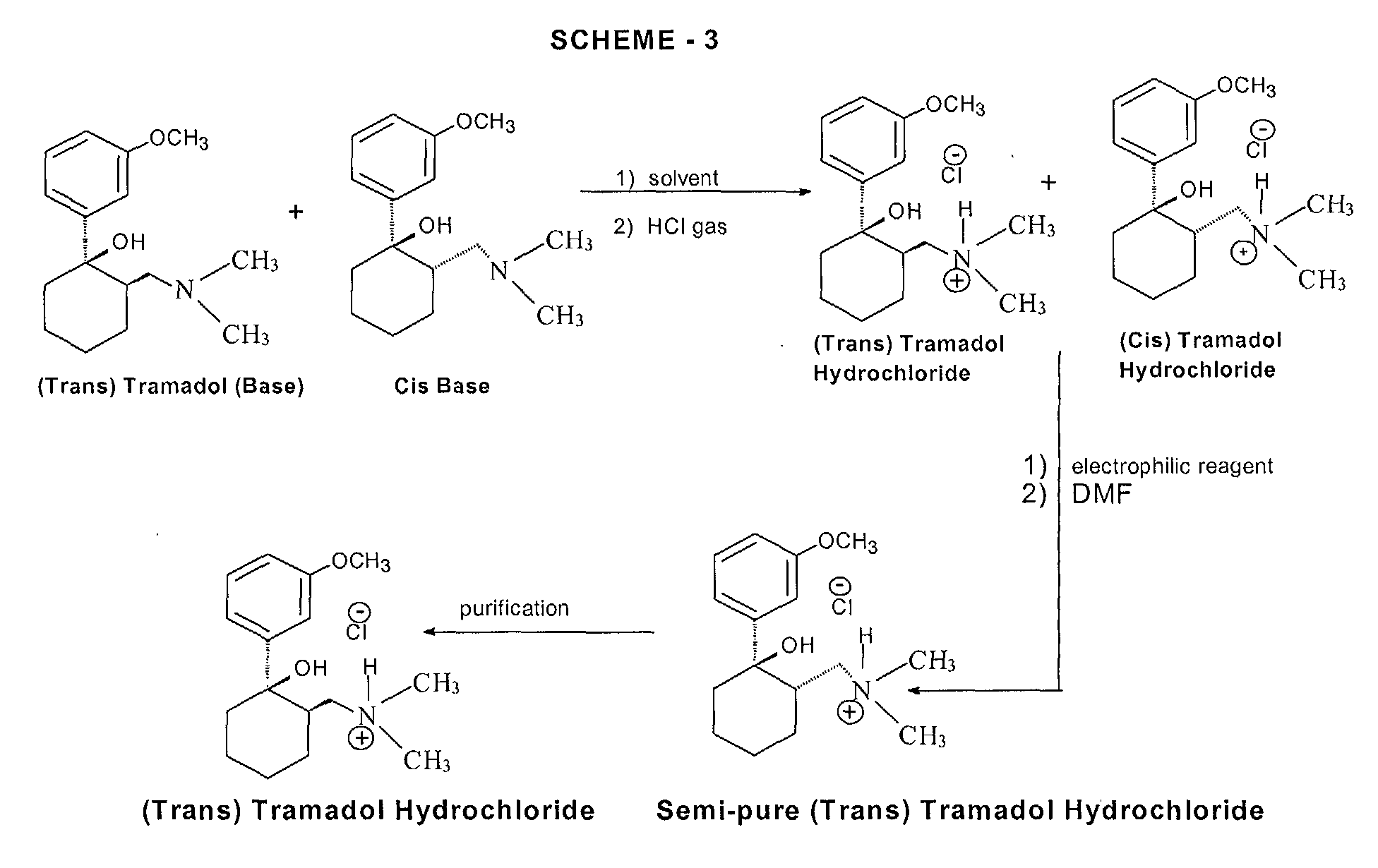
The chemical name for tramadol hydrochloride is (±)cis-2-[(dimethylamino)methyl]-1-(3methoxyphenyl) cyclohexanol hydrochloride
Tramadol (marketed as the hydrochloride salt by Janssen Pharmaceutica as Ultram in the United States, Ralivia by Biovail in Canada and many other companies throughout the world) is a centrally acting synthetic opioid analgesic used to treat moderate to moderately severe pain. The drug has a wide range of applications, including treatment of rheumatoid arthritis, restless legs syndrome, motor neurone disease and fibromyalgia.[citation needed] It was launched and marketed as Tramal by the German pharmaceutical company Grünenthal GmbH in 1977.
Tramadol is a weak μ-opioid receptor agonist, a serotonin releaser and a reuptake inhibitor of norepinephrine. Tramadol is metabolized to O-desmethyltramadol, a significantly more potent μ-opioid agonist. Tramadol and its major metabolite(s) are distinguished from other more potent opioid agonists by relative selectivity for μ-opioid receptors.
Structurally, tramadol closely resembles a stripped down version of codeine. Both codeine and tramadol share the 3-methyl ether group, and both compounds are metabolized along the same hepatic pathway and mechanism to the stronger opioid, phenol agonist analogs. For codeine, this is morphine, and for tramadol, it is the O-desmethyltramadol.
When administered through IV, patients notice very little clinical difference in subjective potency compared to morphine.
Structurally, tapentadol is the closest chemical relative of tramadol in clinical use. Tapentadol is also an opioid, but unlike both tramadol and venlafaxine, tapentadol represents only one stereoisomer and is the weaker of the two, in terms of opioid effect. Both tramadol and venlafaxine are racemic mixtures. Structurally, tapentadol also differs from tramadol in being a phenol, and not an ether. Also, both tramadol and venlafaxine incorporate a cyclohexyl moiety, attached directly to the aromatic, while tapentadol lacks this feature.
-Tramadol.svg)
-Tramadol_gespiegelt.svg)
(1R,2R)-Tramadol (1S,2S)-Tramadol
-Tramadol.svg)
-Tramadol_gespiegelt.svg)
(1R,2S)-Tramadol (1S,2R)-Tramadol
The chemical synthesis of tramadol is described in the literature.[62] Tramadol [2-(dimethylaminomethyl)-1-(3-methoxyphenyl)cyclohexanol] has two stereogenic centers at the cyclohexane ring. Thus, 2-(dimethylaminomethyl)-1-(3-methoxyphenyl)cyclohexanol may exist in four different configurational forms:
The synthetic pathway leads to the racemate (1:1 mixture) of (1R,2R)-isomer and the (1S,2S)-isomer as the main products. Minor amounts of the racemic mixture of the (1R,2S)-isomer and the (1S,2R)-isomer are formed as well. The isolation of the (1R,2R)-isomer and the (1S,2S)-isomer from the diastereomeric minor racemate [(1R,2S)-isomer and (1S,2R)-isomer] is realized by the recrystallization of the hydrochlorides. The drug tramadol is a racemate of the hydrochlorides of the (1R,2R)-(+)- and the (1S,2S)-(–)-enantiomers. The resolution of the racemate [(1R,2R)-(+)-isomer / (1S,2S)-(–)-isomer] was described[63] employing (R)-(–)- or (S)-(+)-mandelic acid. This process does not find industrial application, since tramadol is used as a racemate, despite known different physiological effects[64] of the (1R,2R)- and (1S,2S)-isomers, because the racemate showed higher analgesic activity than either enantiomer in animals[65] and in humans.[66]




On August 20, 2013, Novartis announced in a press release that the FDA had granted breakthrough therapy designation to its experimental agent BYM338 (bimagrumab) for treatment of the rare muscle wasting disease sporadic inclusion body myositis (sIBM).
sIBM is a rare–but increasingly prevalent–disease. It is the most common cause of inflammatory myopathy in people over 50. sIBM has a yearly incidence of 2 to 5 per million adults with a peak at ages 50 to 70, with male predominance. Muscle wasting caused by sIBM is superimposed upon the sarcopenia (degenerative loss of muscle mass) that typically occurs with aging.
read all
bimagrumab
immunoglobulin G1-lambda2, anti-[Homo sapiens ACVR2B (activin
A receptor type IIB, ActR-IIB)], Homo sapiens monoclonal antibody;
gamma1 heavy chain (1-445) [Homo sapiens VH (IGHV1-2*02
(91.80%) -(IGHD)-IGHJ5*01 [8.8.8] (1-115) -IGHG1*03 (CH1 (116-
213), hinge (214-228), CH2 L1.3>A (232), L1.2>A (233) (229-338),
CH3 (339-443), CHS (444-445)) (116-445)], (218-216′)-disulfide with
lambda light chain (1′-217′) [Homo sapiens V-LAMBDA (IGLV2-
23*02 (90.90%) -IGLJ2*01) [9.3.11] (1′-111′) -IGLC2*01 (112′-217′)];
dimer (224-224”:227-227”)-bisdisulfide
myostatin inhibitor
bimagrumab immunoglobuline G1-lambda2, anti-[Homo sapiens ACVR2B
(récepteur type IIB de l’activine A, ActR-IIB)], Homo sapiens
anticorps monoclonal;
chaîne lourde gamma1 (1-445) [Homo sapiens VH (IGHV1-2*02
(91.80%) -(IGHD)-IGHJ5*01 [8.8.8] (1-115) -IGHG1*03 (CH1 (116-
213), charnière (214-228), CH2 L1.3>A (232), L1.2>A (233) (229-
338), CH3 (339-443), CHS (444-445)) (116-445)], (218-216′)-
disulfure avec la chaîne légère lambda (1′-217′) [Homo sapiens
V-LAMBDA (IGLV2-23*02 (90.90%) -IGLJ2*01) [9.3.11] (1′-111′) –
IGLC2*01 (112′-217′)]; dimère (224-224”:227-227”)-bisdisulfure
inhibiteur de la myostatine
inmunoglobulina G1-lambda2, anti-[Homo sapiens ACVR2B
(receptor tipo IIB de la activina A, ActR-IIB)], anticuerpo monoclonal
de Homo sapiens;
cadena pesada gamma1 (1-445) [Homo sapiens VH (IGHV1-2*02
(91.80%) -(IGHD)-IGHJ5*01 [8.8.8] (1-115) -IGHG1*03 (CH1 (116-
213), bisagra (214-228), CH2 L1.3>A (232), L1.2>A (233) (229-338),
CH3 (339-443), CHS (444-445)) (116-445)], (218-216′)-disulfuro con
la cadena ligera lambda (1′-217′) [Homo sapiens V-LAMBDA
(IGLV2-23*02 (90.90%) -IGLJ2*01) [9.3.11] (1′-111′) -IGLC2*01
(112′-217′)]; dímero (224-224”:227-227”)-bisdisulfuro
inhibidor de la miostatina
1356922-05-8
Heavy chain / Chaîne lourde / Cadena pesada
QVQLVQSGAE VKKPGASVKV SCKASGYTFT SSYINWVRQA PGQGLEWMGT 50
INPVSGSTSY AQKFQGRVTM TRDTSISTAY MELSRLRSDD TAVYYCARGG 100
WFDYWGQGTL VTVSSASTKG PSVFPLAPSS KSTSGGTAAL GCLVKDYFPE 150
PVTVSWNSGA LTSGVHTFPA VLQSSGLYSL SSVVTVPSSS LGTQTYICNV 200
NHKPSNTKVD KRVEPKSCDK THTCPPCPAP EAAGGPSVFL FPPKPKDTLM 250
ISRTPEVTCV VVDVSHEDPE VKFNWYVDGV EVHNAKTKPR EEQYNSTYRV 300
VSVLTVLHQD WLNGKEYKCK VSNKALPAPI EKTISKAKGQ PREPQVYTLP 350
PSREEMTKNQ VSLTCLVKGF YPSDIAVEWE SNGQPENNYK TTPPVLDSDG 400
SFFLYSKLTV DKSRWQQGNV FSCSVMHEAL HNHYTQKSLS LSPGK 445
Light chain / Chaîne légère / Cadena ligera
QSALTQPASV SGSPGQSITI SCTGTSSDVG SYNYVNWYQQ HPGKAPKLMI 50
YGVSKRPSGV SNRFSGSKSG NTASLTISGL QAEDEADYYC GTFAGGSYYG 100
VFGGGTKLTV LGQPKAAPSV TLFPPSSEEL QANKATLVCL ISDFYPGAVT 150
VAWKADSSPV KAGVETTTPS KQSNNKYAAS SYLSLTPEQW KSHRSYSCQV 200
THEGSTVEKT VAPTECS 217
Disulfide bridges location / Position des ponts disulfure / Posiciones de los puentes disulfuro
Intra-H 22-96 142-198 259-319 365-423
22”-96” 142”-198” 259”-319” 365”-423”
Intra-L 22′-90′ 139′-198′
22”’-90”’ 139”’-198”’
Inter-H-L 218-216′ 218”-216”’
Inter-H-H 224-224” 227-227”
N-glycosylation sites / Sites de N-glycosylation / Posiciones de N-glicosilación
H CH2 N84.4
Bimagrumab
http://www.who.int/medicines/publications/druginformation/innlists/PL108_Final.pdf
Novartis announced that the US Food and Drug Administration (FDA) has granted breakthrough therapy designation to BYM338 for sporadic inclusion body myositis (sIBM). This designation is based on the results of a phase 2 proof-of-concept study that showed BYM338 substantially benefited patients with sIBM compared to placebo.
read all at
Novartis receives FDA breakthrough therapy designation for BYM338 (bimagrumab) for sporadic inclusion body myositis (sIBM)
• Designation highlights potential of BYM338 to address an unmet medical need in a serious disease
• If approved, BYM338 has the potential to be the first treatment for sIBM patients
• BYM338 is the third Novartis investigational treatment this year to receive a breakthrough therapy designation by the FDA, highlighting Novartis’ leadership in the industry in breakthrough therapy designations
Bimagrumab (BYM338) is a human monoclonal antibody developed by Novartis to treat pathological muscle loss and weakness. On August 20, 2013 it was announced that bimagrumab was granted breakthrough therapy designation for sporadic inclusion body myositis(sIBM) by US Food and Drug Administration.[1]
World Health Organization (2012). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 108″ (PDF). WHO Drug Information 26(4)

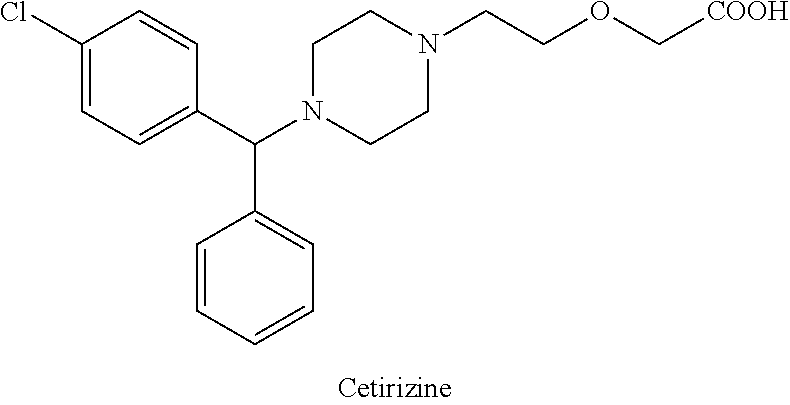
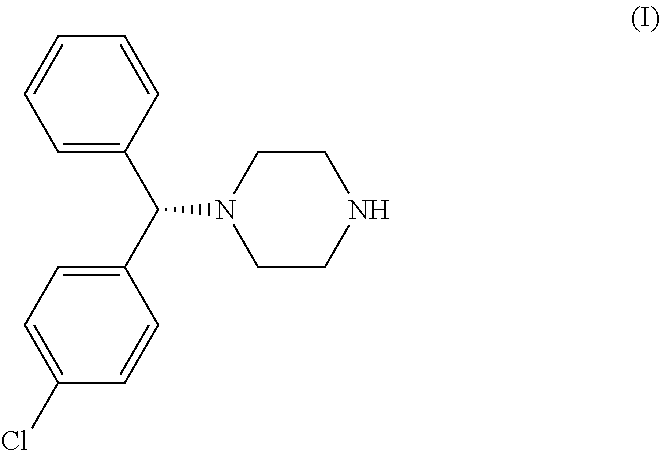
LEVOCETRIZINE



 |
| (Figure 2) |
The synthesis of Cetrizine begins by reducing molecule 33 with a catecholborane. This reaction yields molecule 34, which is then treated with tetraflouroboric acid and reacted with an amine, compound 35. In order to remove the chromium group, the compound is refluxed in pyridine and undergoes an acid hydrolysis. This results in a yield of cetrizine.
Identification of all chirality centers:
Stereogenic centers are carbon atoms that are bonded to 4 groups. Tetrahedral stereogenic centers are stereogenic centers that are not only bonded to 4 groups but are more importantly bonded to 4 different groups. If a molecule contains 1 tetrahedral stereogenic center it is said to be chiral (nonsuperimposable on its mirror image).If a given compound contains more than 1 stereogenic center it must be further analyzed to determine if it is chiral or achiral(superimosable on its mirror image). The carbon atom bonded to the phenyl groups was found to be a tetrahedral stereogenic center. Therefore,xyzal,which was found to contain only 1 tetrahedral stereogenic center is generally considered a chiral compound because it meets the requirements of chirality and does not have a plane of symmetry that superimposes one half of the molecule on the other and is not super imposable on its mirror image.
Spectral data for Xyzal (IR and NMR):


– See more at: http://worlddrugtracker.blogspot.in/#sthash.6Ndn04WU.dpuf

ABSTRACT:
The prostaglandins are a family of lipids, originally discovered over 30 years ago in human seminal fluid, which have since been found not only to have a wide variety of striking pharmacological actions, but also to be present in many if not all mammalian tissues. They have an unusual chemical structure, being 20-carbon fatty acids derived enzymically from the essential fatty acids by cyclization and oxidation.
read all at
http://www.pharmatutor.org/articles/prostaglandin-useful-strategy-new-drug-development-review


This unassuming weed is currently the source for of the anticancer drug ingenol © Floral Images/Alamy
US scientists have developed the first efficient and scalable route for the total synthesis of ingenol – a plant-derived diterpenoid used to treat precancerous skin legions. The work offers cheaper and faster production of the drug than the current, inefficient plant extraction route, and could pave the way for the chemical synthesis of many other complex natural compounds.
read all at
http://www.rsc.org/chemistryworld/2013/08/total-synthesis-outshines-biotech-anticancer-drug-ingenol
.
The desire for a dramatic increase in process understanding over
the past eight years has left industry leaders and Global Health
Agencies searching for a more relevant model for developing process and
drug substance understanding. Most recently, the Internationa l Conference on
Harmonization (ICH) has been drafting the ICH guideline q11 on the development and manufacture of drug substances, which takes a considerable step forward, offering sponsors greater flexibility in the definition and selection of Regulatory Starting Materials (RSM).
http://www.triphasepharmasolutions.com/Regulatory%20API%20Starting%20Materials%20Article.pdf

A new therapeutic approach can take advantage of cancer cells’ need to repair double-strand breaks in DNA, in order to overcome the tumour’s resistance to chemotherapy (Science Transl. Medicine). According to the research group headed by Hans Christian from University Cologne, mutations in the ATM gene protect cancer cells from cell death during chemotherapy. ATM is instrumental in initiating DNA repair and inducing cell death when repair is not possible in curse of the so-called DNA damage response (DDR). http://www.eurobiotechnews.eu/news/news/2013-02/double-strike-against-cancer.html
| ThromboGenics’ Jetrea receives NICE approval for eye condition treatment |
| Biopharmaceutical company ThromboGenics’ Jetrea has received approval from UK’s National Institute for Health and Care Excellence (NICE) in the treatment of some adults with vitreomacular traction (VMT), a rare eye condition… |
Ocriplasmin (trade name Jetrea) is a recombinant protease with activity against fibronectin and laminin, components of the vitreoretinal interface. It is used for treatment of symptomatic vitreomacular adhesion, for which it received FDA approval on 17 October 2012. It works by dissolving the proteins that link the vitreous to the macula, resulting in posterior detachment of the vitreous from theretina.[1]
