
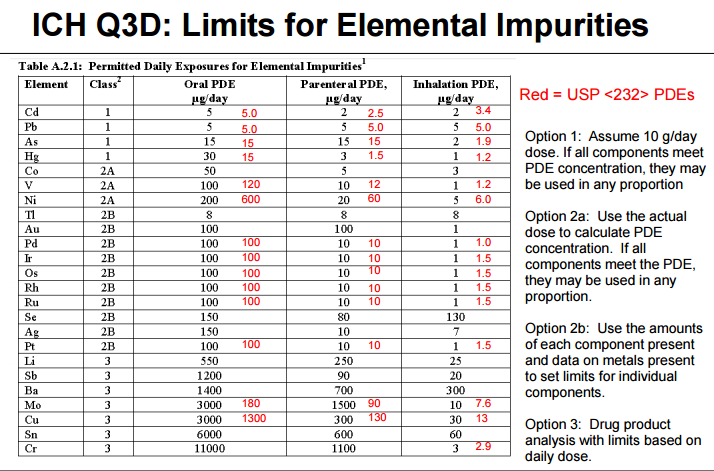
On January 1, 2018, new guidelines regarding elemental impurities in brand and generic drug products went into effect. Elemental impurities, such as arsenic and lead, pose toxicological risks to patients without providing any therapeutic benefit. These impurities may be present in drug products from a variety of sources, such as interactions with equipment during the drug manufacturing process.
FDA, together with other organizations, such as the International Council for Harmonisation (ICH) and the U.S. Pharmacopeial Convention (USPC), have engaged in long-standing efforts to best protect patients from the risks posed by elemental impurities by developing limits for their amounts in drug products, and standardized approaches to use in determining the amount of elemental impurities in these products.
As of January 1, 2018:
- All new and existing NDAs and ANDAs for drug products with an official USP monograph are required to meet the requirements in USP General Chapters <232> and <233> for the control of elemental impurities.
- Applicants submitting NDAs and ANDAs for drug products without a USP monograph are expected to follow the recommendations in the ICH Q3D Elemental Impurities
 guideline.
guideline.
Questions and Answers on Elemental Impurities:
Why were these guidelines developed, and why are they important?
Heavy metal elemental impurities pose serious risks to patients without providing a benefit. Modern methods provide better analytical tests to detect elemental impurities, which in turn, will help protect patients by ensuring approved products have safe levels of these impurities. The ICH guidelines and USP General Chapters <232>Elemental Impurities—Limits are focused on establishing Permitted Daily Exposures (PDEs) for elemental impurities in drug products. USP General Chapter <233>Elemental Impurities—Procedures describes analytical approaches for the detection of elemental impurities. The analytical approaches described in <233> are based on modern analytical capabilities, replace the outdated tests in the deleted USP General Chapter <231> Heavy Metals, and allow us to more precisely measure impurities to ensure safe levels. FDA, ICH, USP, and industry experts worked together to develop the new standards that are in alignment and help ensure high quality medicines.
How has FDA been supporting industry to implement the requirements?
FDA, ICH, and USP have all engaged with brand and generic drug manufacturers to support implementation of these requirements. These requirements are the result of long-standing efforts, and both ICH and USP included industry participants on their expert panels that developed these standards. With that input, an implementation date was identified that provided firms with substantial time to verify their operations met the requirements.
In June 2016, FDA published a draft guidance, Elemental Impurities in Drug Products, to provide recommendations regarding the control of elemental impurities of human drug products. The draft guidance encouraged the early adoption of ICH Q3D guidelines and USP General Chapters <232> and <233> before the January 1, 2018 implementation date. FDA has also presented on this topic at conferences, including at a two-day ICH Q3D regional workshop it hosted in August 2016 1. These outreach efforts have supported efforts by industry to perform the risk assessments needed to implement the new guidelines in order to have complete, approvable applications. On an application-specific level, FDA began noting this requirement in complete response letters to applicants that contained quality deficiencies in Spring of 2017.
What should companies do if they have questions about elemental impurity standards?
Companies that have quality questions regarding elemental impurities and their applications should contact the Regulatory Business Process Manager (RBPM) in the Office of Program and Regulatory Operations, Office of Pharmaceutical Quality for their application. Applications that do not meet the elemental impurity guidelines are unable to be approved and applicants may receive a request for the information from the FDA in the form of an Information Request or a Complete Response letter. Firms should submit information on their elemental impurity risk assessments to FDA as soon as they are able, rather than waiting for a request from FDA, in order to minimize the impact on review and approval timeframes. The following resource may help applicants understand the process moving forward depending on where they are in the review process.
What is the International Council for Harmonisation?
ICH, first created in 1990 by regulatory agencies and both brand and generic drug manufacturing associations from the United States, Europe, and Japan, was established to facilitate international collaboration, and has been successful in standardizing and elevating drug development practices throughout the world. ICH’s mission helps to increase patient access to safe, effective, and high quality pharmaceuticals, and to ensure that pharmaceuticals are developed and registered efficiently. International harmonization of regulatory standards means that pharmaceutical manufacturers and developers will be held to the same standards in different markets (countries), which will make the development and delivery of quality pharmaceuticals to the public more timely and efficient. The ICH Website includes training modules on implementation of the Q3D elemental impurity guidelines.
What is the U.S. Pharmacopeia Convention?
The United States Pharmacopeia Convention (USPC) is a private non-profit organization that develops public standards related to pharmaceutical quality. USP General Chapters <232>Elemental Impurities—Limits, and, <233>Elemental Impurities—Procedures are applicable to compendial drug products as per Federal Food, Drug, and Cosmetic Act Sec. 201(j), and Sec. 501(b). USP’s website offers information regarding the history of actions they have taken on elemental impurities, as well as other FAQ.
1 Other presentations include the Drug Information Association’s CMC Workshop 2015, the Consumer Healthcare Products Association’s 2015 Regulatory, Scientific & Quality Conference, the Product Quality Research Institute (PQRI) / USP Workshop on ICH Q3D Elemental Impurities Requirements, the Generic Pharmaceutical Association (now Association of Affordable Medicines) CMC Workshop, the USP Excipients Stakeholder Forum, the PQRI/USP Workshop on Implementation Status of ICH Q3D, and the PQRI/USP Workshop on ICH Q3D Elemental Impurities Requirements – Recent Experience and Plans for Full Implementation in 2018
Elemental Impurities
Efforts in this area are currently focused on three fronts:
-
Finalization of risk assessments to ensure compliance with the ICH Q3D guideline for all products supplied to those markets having implemented ICH Q3D and to the date for implementation
-
Continued development of ICH Q3D dermal limits
-
Removal of the heavy metals limit test USP <231>

Marketed Product Compliance
When it was published at the end of 2014, ICH Q3D
(1) provided a 3 year moratorium in relation to established products, meaning that all such products would have to demonstrate compliance with the guideline at the end of 2017. Many involved will testify to the Herculean effort required to complete this within large organizations where hundreds if not thousands of products were within scope. What has been the outcome? Informal feedback within the industry is that aside from a small number of products, organizations have found that the vast majority of products assessed require no additional control measures because they already have appropriate quality control measures.
Elemental Impurities within Excipients
The ICH Q3D guideline describes how a risk-based approach to the control of elemental impurities in drug products can be taken, highlighting within this that assessments should be data-driven. Options in terms of data include both data generated specific to a drug product and published data. In 2015 the U.S. Food and Drug Administration (FDA) and the European International Pharmaceutical Excipient Council (IPEC) jointly published the outcome of a focused study on some 200 excipient samples covering a range of excipients. This concluded that the overall risk associated with excipients, including those that are mined, was relatively low, especially when typical proportions in formulated drug products were considered. With the express aim of building upon this initial study, a consortium of pharmaceutical companies has established a database to collate the results of analytical studies of the levels of elemental impurities within pharmaceutical excipients. This database currently includes the results of over 25 000 elemental determinations for over 200 different excipients and represents the largest known, and still rapidly expanding, collection of data of this type.

A recently published analysis of the database
(2) examined a series of aspects, including data coverage as well as impurity levels and variability (across supplier/grade, etc.). The database includes results from multiple analytical studies for many of the excipients and thus can give a clear indication of both excipient supplier and batch-to-batch variability as well as any variability associated with the different testing organizations and methods employed. The results are telling. Critically, the data confirm the findings of earlier, smaller FDA–IPEC studies showing that elemental impurity concentrations in excipients, including mined excipients, are generally low and when used in typical proportions in formulated drug products are unlikely to pose a significant patient safety risk.
The database is now in active use within member organizations, providing real evidence in support of holistic ICH Q3D risk assessments and in the future potentially significantly reducing the need for testing. However, it is necessary to recognize that there was a sense that mined excipients could still present a risk over the long term. That variability in elemental impurity levels within mined excipients will vary over time, and further data will be required. There is therefore a need for continued collaboration between the pharmaceutical industry and excipient manufacturers.
It is interesting to reflect that had such studies been conducted ahead of finalization of ICH Q3D, it is possible that it would have allowed us to eliminate concerns about elemental impurities, at least for some low-risk excipients Another study could have achieved the same outcome for manufacturing equipment.
Removal of Heavy Metals Testing
Perhaps our biggest challenge as an industry in this area relates to the potential to remove existing empirical testing for elemental impurities using the wet-chemistry heavy metals limit test because of differences in the global regulatory landscape. In the case of the United States Pharmacopeia (USP), this takes the form of the now-deleted USP Chapter <231>.
On the basis of the time scale for implementation of ICH Q3D, most organizations are well-advanced in terms of the risk assessment of current products, as described above. In the clear majority of cases, this successfully demonstrates that the heavy metals test does not provide any additional control for elemental impurities. On this basis, it should therefore be possible to remove the heavy metals limit test, of which USP <231> is the most prevalent example.
The situation in the U.S. is that removal is relatively straightforward, as the test has already been removed from the USP. A statement to confirm completion of an elemental impurity risk assessment is then provided in the product annual update. Elsewhere, the situation is more challenging. In Europe there is no definitive position, but filing a simple show-and-tell type 1A variation seems to provide a pathway. Thereafter, the situation is considerably more complex.
In Japan, the equivalent of the USP <231> test has been retained in the Japanese Pharmacopeia (JP). Consequently, removing the test from an existing product (one where a monograph is published and it includes such a test) may require submitting a product-specific request to revise the individual monograph. It is also anticipated that removal of the test from approved but not monographed products will also require a post-approval change submission.
In China, the Chinese Pharmacopeia (CP) will retain the test until at least 2020, and the indication is that the test should still be performed where registered.
Outside of ICH regions, the situation is still more complicated. Given the prevalent position of the USP in many countries, API and product specifications often include USP <231>. However, this test no longer exists! The challenge then concerns whether the test can be removed and the specification revised, and if so, how this should be done. The scale of this is significant, especially if a formal variations procedure is needed. One apparent option is to continue testing, but even this is complicated, as it is not clear how one could continue to use a test that no longer exists in the USP. Some organizations have even considered developing a “USP <231>-like” test.
Clearly, organizations do not want to continue to use an empirical test when a risk assessment has shown that it adds no value, but at present there is no obvious way to resolve this conundrum for globally marketed products until significant harmonization in compendial test requirements is achieved.
REFERENCES
1 Guideline for Elemental Impurities Q3D, Current Step 4 version, dated Dec 16, 2014.
2 Boetzel, R.; Ceszlak, A.; Day, C.An Elemental Impurities Excipient Database: A Viable Tool for ICH Q3D Drug Product Risk Assessment. J. Pharm. Sci. 2018, DOI: 10.1016/j.xphs.2018.04.009
//////////Elemental Impurities, ICH Q3D, USP













 , Hideyoshi Tsuchiya, Mitsuru Teramoto, Naoki Yajima, Masayuki Kurokawa, Kunio Minamizono, Naoki Tsuchiya, Yoshiaki Kato, Yoshinori Sato, and Masahiko Dohi
, Hideyoshi Tsuchiya, Mitsuru Teramoto, Naoki Yajima, Masayuki Kurokawa, Kunio Minamizono, Naoki Tsuchiya, Yoshiaki Kato, Yoshinori Sato, and Masahiko Dohi



























.png)





 LinkedIn
LinkedIn Facebook
Facebook Twitter
Twitter GooglePlus
GooglePlus