Green Chem., 2017, Advance Article
DOI: 10.1039/C6GC03123D, Paper
Maurice A. Marsini, Frederic G. Buono, Jon C. Lorenz, Bing-Shiou Yang, Jonathan T. Reeves, Kanwar Sidhu, Max Sarvestani, Zhulin Tan, Yongda Zhang, Ning Li, Heewon Lee, Jason Brazzillo, Laurence J. Nummy, J. C. Chung, Irungu K. Luvaga, Bikshandarkoil A. Narayanan, Xudong Wei, Jinhua J. Song, Frank Roschangar, Nathan K. Yee, Chris H. Senanayake
A convergent and robust synthesis of a developmental CCR1 antagonist is described using continuous flow technology
http://pubs.rsc.org/en/Content/ArticleLanding/2017/GC/C6GC03123D?utm_source=feedburner&utm_medium=feed&utm_campaign=Feed%3A+rss%2FGC+%28RSC+-+Green+Chem.+latest+articles%29#!divAbstract
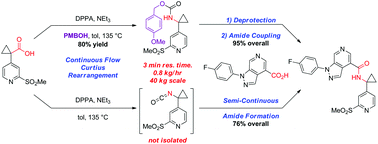
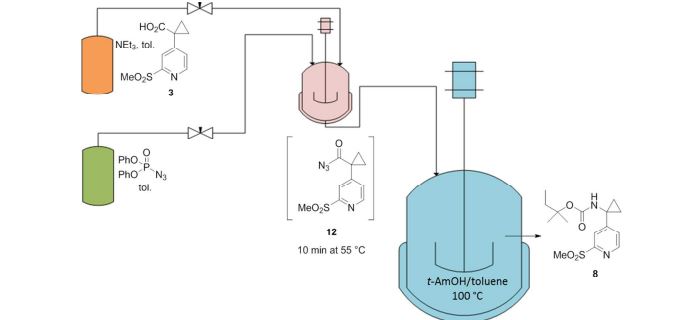
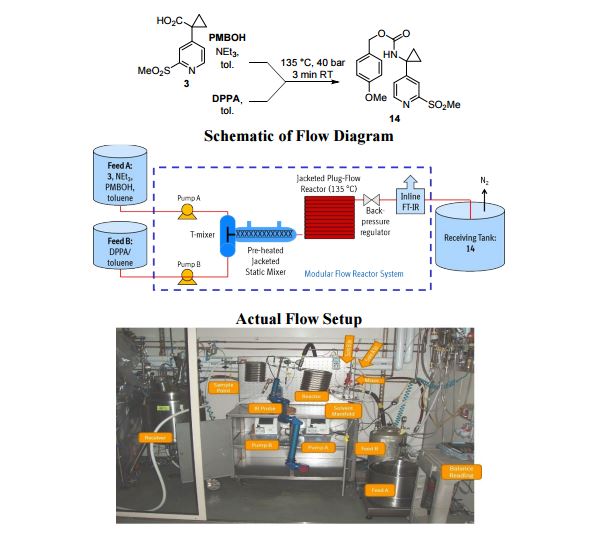
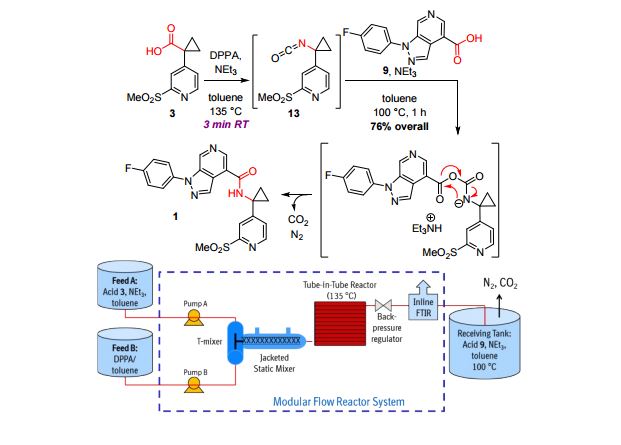
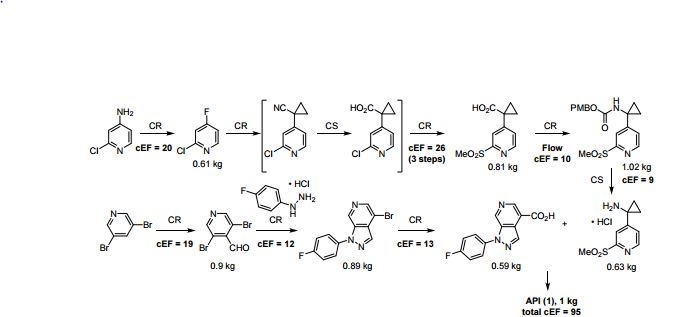
A convergent, robust, and concise synthesis of a developmental CCR1 antagonist is described using continuous flow technology. In the first approach, following an expeditious SNAr sequence for cyclopropane introduction, a safe, continuous flow Curtius rearrangement was developed for the synthesis of a p-methoxybenzyl (PMB) carbamate. Based on kinetic studies, a highly efficient and green process comprising three chemical transformations (azide formation, rearrangement, and isocyanate trapping) was developed with a relatively short residence time and high material throughput (0.8 kg h−1, complete E-factor = ∼9) and was successfully executed on 40 kg scale. Moreover, mechanistic studies enabled the execution of a semi-continuous, tandem Curtius rearrangement and acid–isocyanate coupling to directly afford the final drug candidate in a single, protecting group-free operation. The resulting API synthesis is further determined to be extremely green (RPG = 166%) relative to the industrial average for molecules of similar complexity.
Development of a concise, scalable synthesis of a CCR1 antagonist utilizing a continuous flow Curtius rearrangement
Maurice A. Marsini,*a Frederic G. Buono,*a Jon C. Lorenz,a Bing-Shiou Yang,a Jonathan T. Reeves,a Kanwar Sidhu,a Max Sarvestani,a Zhulin Tan,a Yongda Zhang,a Ning Li,a Heewon Lee,a Jason Brazzillo,a Laurence J. Nummy,a J. C. Chung,a Irungu K. Luvaga,a Bikshandarkoil A. Narayanan,a Xudong Wei,a Jinhua J. Song,a Frank Roschangar,a Nathan K. Yeea and Chris H. Senanayakea
Green Chem., 2017, Advance Article
DOI: 10.1039/C6GC03123D

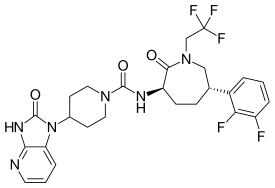
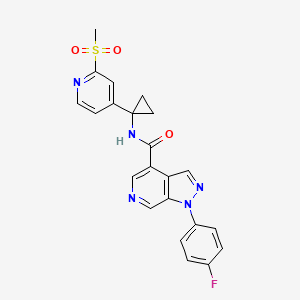
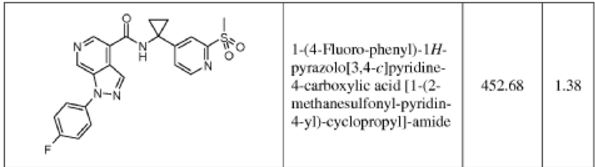
1-(4-fluorophenyl)-N-(1-(2-(methylsulfonyl)pyridin-4-yl)cyclopropyl)-1H-pyrazolo[3,4- c]pyridine-4-carboxamide
1-(4-fluorophenyl)-N-(1-(2-(methylsulfonyl)pyridin-4-yl)cyclopropyl)-1H-pyrazolo[3,4- c]pyridine-4-carboxamide
m.p. = 140-144 °C;
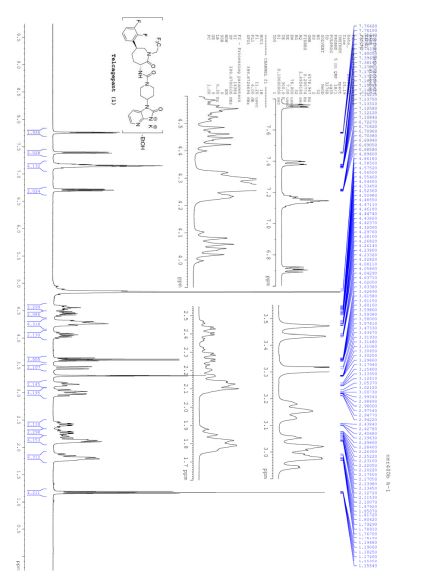
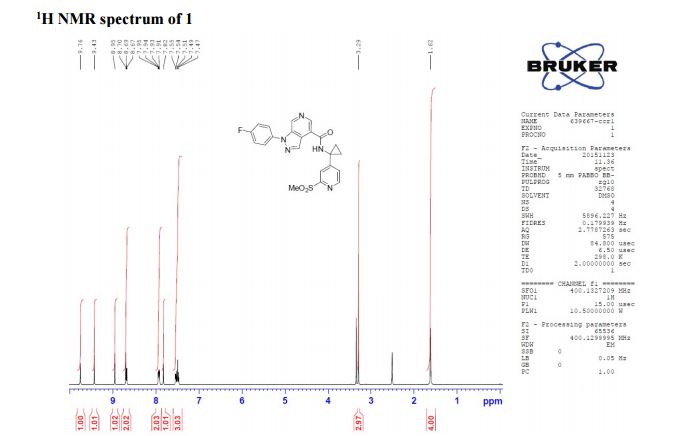
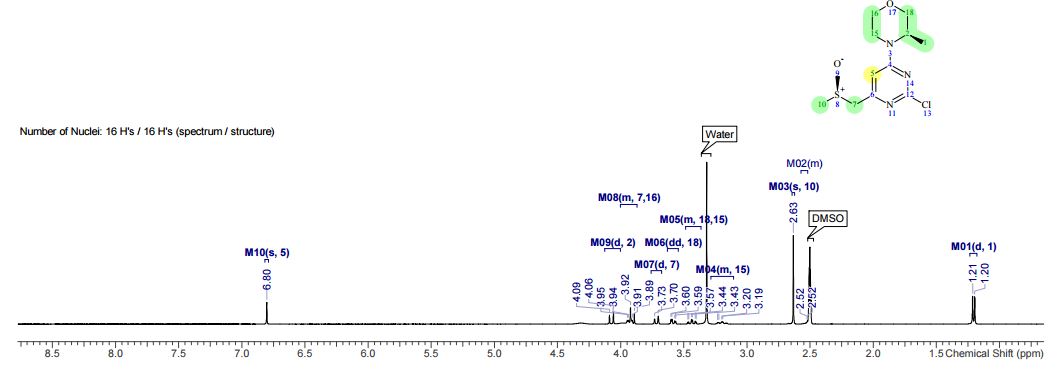
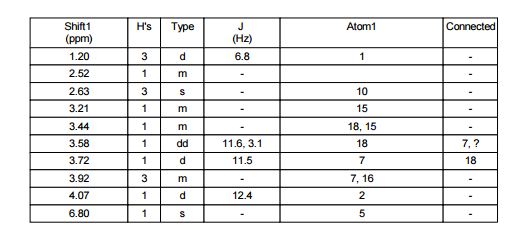
1H NMR (400 MHz, CDCl3) δ 9.76 (s, 1H), 9.43 (s, 1H), 8.95 (s, 1H), 8.70 (s, 1H), 8.68 (d, J = 5.2 Hz, 1H), 7.93 (s, J1 = 8.8 Hz, J2 = 4.7 Hz, 1H), 7.82 (s, 1H), 7.54 (d, J = 4.1 Hz, 1H), 7.49 (t, J = 8.7 Hz, 1H), 3.29 (s, 3H), 1.61 (bs, 4H);
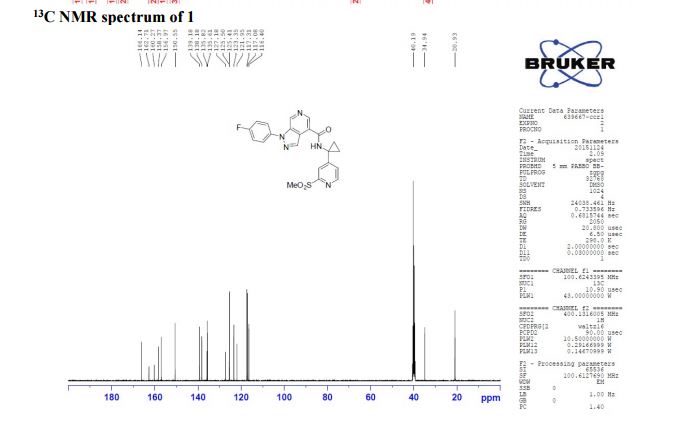
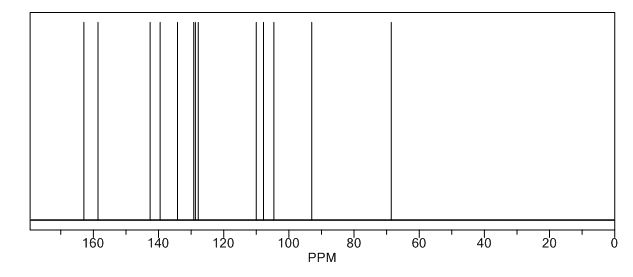
13C NMR (100 MHz, CDCl3) δ 166.1, 162.7, 160.3, 158.4, 156.9, 150.6, 139.2, 138.2, 135.8, 135.6, 125.4 (d, JC-F = 8.8 Hz), 123.3, 121.9, 117.2 (d, JC-F = 23.1 Hz), 116.4, 40.2, 34.9, 20.9;
HRMS: calcd for C22H19FN5O3S [M + H+ ]: 452.1187. Found: 452.1189.
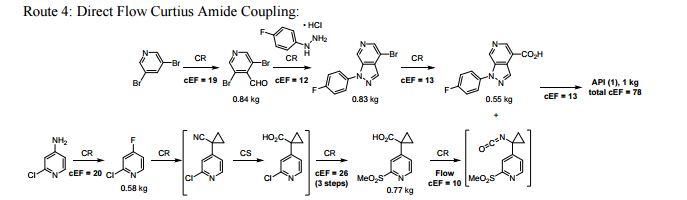
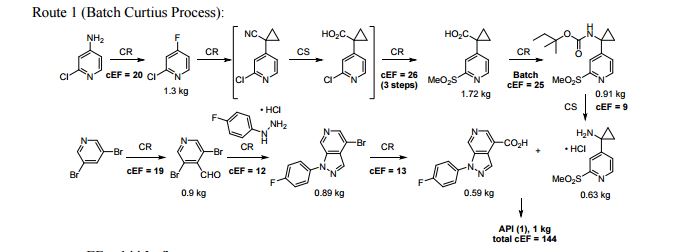
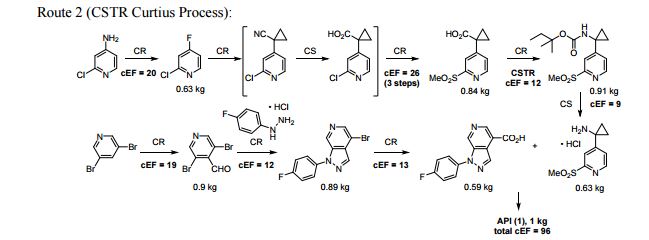
//////////BI-638683, BI 638683, CCR1 antagonist, 295298-26-8, US2012270870, Boehringer Ingelheim Pharmaceuticals, phase 1
CS(=O)(=O)c1nccc(c1)C2(CC2)NC(=O)c5cncc3c5cnn3c4ccc(F)cc4
| Molecular Formula: |
C22H18FN5O3S |
| Molecular Weight: |
451.476 g/mol |
cas 295298-26-8
US2012270870
maybe BI-638683, not sure
In September 2010, a randomized, double-blind, placebo-controlled, phase I study (NCT01195688; 1279.1; 2010-021187-15) was initiated in healthy male volunteers (expected n = 64) in Germany, to assess the safety, pharmacokinetics and pharmacodynamics of BI-638683. The study was completed in December 2010 . In June 2014, data were presented at the EULAR 2014 Annual Meeting in Paris, France. A dose of 75-mg showed maximal inhibition of mRNA expression of the four-CC chemokine receptor type-I dependent marker genes. chemokine ligand -2 and Peroxisome proliferator-activated receptor gamma-mRNAs by doses of 300 mg and higher, and for Ras-related protein rab-7b mRNA by doses of 500 mg and higher
Boehringer Ingelheim was developing BI-638683, a CCR1 antagonist, for the potential oral treatment of rheumatoid arthritis. A phase I trial was completed in December 2010 . Phase I data was presented in June 2014
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011049917&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
Chemotactic Cytokine Receptor 1 (CCRl) belongs to a large family (>20) of chemotactic cytokine (chemokine) receptors that interact with specific chemokines (>50) to mediate leukocyte trafficking, granule exocytosis, gene transcription, mitogenic effects and apoptosis. Chemokines are best known for their ability to mediate basal and inflammatory leukocyte trafficking. The binding of at least three chemokines (MIP-1 alpha/CCL3, MCP3/CCL7 and RANTES/CCL5) to CCRl is responsible for the trafficking of monocytes, macrophages and THl cells to inflamed tissues of rheumatoid arthritis (RA) and multiple sclerosis (MS) patients (Trebst et al. (2001) American J of Pathology 159 p. 1701). Macrophage inflammatory protein 1 alpha (MIP-1 alpha), macrophage chemoattractant protein 3 (MCP-3) and regulated on activation, normal T-cell expressed and secreted (RANTES) are all found in the CNS of MS patients, while MIP-1 alpha and RANTES are found in the CNS in the experimental autoimmune encephalomyelitis (EAE) model of MS (Review: Gerard
and Rollins (2001) Nature Immunology). Macrophages and Thl cells in the inflamed synovia of RA patients are major producers of MIP-1 alpha and RANTES, which continuously recruit leukocytes to the synovial tissues of RA patients to propagate chronic inflammation (Volin et al. (1998) Clin. Immunol. Immunopathology; Koch et al. (1994) J. Clin. Investigation; Conlon et al. (1995) Eur. J. Immunology). Antagonizing the interactions between CCR1 and its chemokine ligands is hypothesized to block chemotaxis of monocytes, macrophages and Thl cells to inflamed tissues and thereby ameliorate the chronic inflammation associated with autoimmune diseases such as RA and MS.
Evidence for the role of CCR1 in the development and progression of chronic inflammation associated with experimental autoimmune encephalitis (EAE), a model of multiple sclerosis, is based on both genetic deletion and small molecule antagonists of CCR1. CCR1 deficient mice were shown to exhibit reduced susceptibility (55% vs. 100%) and reduced severity (1.2 vs. 2.5) of active EAE (Rottman et al. (2000) Eur. J. Immunology). Furthermore, administration of small molecule antagonist of CCR1, with moderate affinity (K; = 120 nM) for rat CCR1, was shown to delay the onset and reduce the severity of EAE when administered intravenously (Liang et al. (2000) /. Biol. Chemistry). Treatment of mice with antibodies specific for the CCR1 ligand MIP- 1 alpha have also been shown to be effective in preventing development of acute and relapsing EAE by reducing the numbers of T cells and macrophages recruited to the CNS (Karpus et al. (1995) /. Immunology; Karpus and Kennedy (1997) /. Leukocyte Biology). Thus, at least one CCR1 ligand has been demonstrated to recruit leukocytes to the CNS and propagate chronic inflammation in EAE, providing further in vivo validation for the role of CCR1 in EAE and MS.
In vivo validation of CCR1 in the development and propagation of chronic inflammation associated with RA is also significant. For example, administration of a CCR1 antagonist in the collagen induced arthritis model (CIA) in DBA/1 mice has been shown to be effective in reducing synovial inflammation and joint destruction (Plater-Zyberk et al. (1997) Immunology Letters). Another publication described potent antagonists of murine CCR1 that reduced severity (58%) in LPS-accelerated collagen-induced arthritis (CIA), when administered orally {Biorganic and Medicinal Chemistry Letters 15, 2005, 5160-5164). Published results from a Phase lb clinical trial with an oral CCRl antagonist demonstrated a trend toward clinical improvement in the absence of adverse side effects (Haringman et al. (2003) Ann. Rheum. Dis.). One third of the patients achieved a 20% improvement in rheumatoid arthritis signs and symptoms (ACR20) on day 18 and CCRl positive cells were reduced by 70% in the synovia of the treated patients, with significant reduction in specific cell types including 50% reduction in CD4+ T cells, 50% reduction in CD8+ T cells and 34% reduction in macrophages.
Studies such as those cited above support a role for CCRl in MS and RA and provide a therapeutic rationale for the development of CCRl antagonists.













 , Bradley Adams, Helen Benson, Julie Demeritt†∥, Steven McKown, Keith Mulholland, Amy Robertson, Paul Siedlecki, Paula Tomlin, and Kevin Vare
, Bradley Adams, Helen Benson, Julie Demeritt†∥, Steven McKown, Keith Mulholland, Amy Robertson, Paul Siedlecki, Paula Tomlin, and Kevin Vare



 gene.com
gene.com