Route to Benzimidazol-2-ones via Decarbonylative Ring Contraction of Quinoxalinediones: Application to the Synthesis of Flibanserin, A Drug for Treating Hypoactive Sexual Desire Disorder in Women and Marine Natural Product Hunanamycin Analogue
† Division of Organic Chemistry, CSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pune 411008, India
‡ Academy of Scientific and Innovative Research (AcSIR), New Delhi 110 025, India
ACS Omega, 2017, 2 (8), pp 5137–5141
DOI: 10.1021/acsomega.7b00819
ACS AuthorChoice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes.
INTRODUCTION
Benzimidazol-2-ones 1 are an important class of heterocycles and a privileged scaffold in medicinal chemistry. They consist of cyclic urea fused with the aromatic backbone, which can potentially interact in a biological system by various noncovalent interactions such as hydrogen bonding and π stacking. Benzimidazolone derivatives exhibit a wide range of biological activities, and they are useful in treating various diseases including cancer, type II diabetes, central nervous system disorders, pain management, and infectious disease.1 Selected compounds embedded with a benzimidazol-2-one moiety along with their use are captured in Figure 1. It is worth mentioning that oxatomide drug with a benzimidazol-2-one core was approved for marketing a few years ago.2a Very recently, US Food and Drug Administration approved a new drug called flibanserin for the treatment of hypoactive sexual desire disorder (HSDD) in females, which contains benzimidazol-2- one motif.2b
CONCLUSIONS
We have developed a mild and new protocol for the synthesis of benzimidazol-2-ones from quinoxalinediones through decarbonylation. The present methodology can be an addition to the toolbox to prepare benzimidazolones, and it will be useful in medicinal chemistry, particularly, late-stage functionalization of natural products, drug scaffolds, or an intermediate containing quinoxaline-2,3-diones. As direct application of this method, we have successfully developed a new route for the synthesis of recently approved drug flibanserin and a urea analogue of antibiotic natural product hunanamycin A. Later application demonstrates the utility of the present method in late-stage functionalization
Synthesis of 1-(2-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)ethyl)-1,3-dihydro-2Hbenzo[d]imidazol-2-one (Flibanserin)
Flibanserin hydrochloride as white solid.
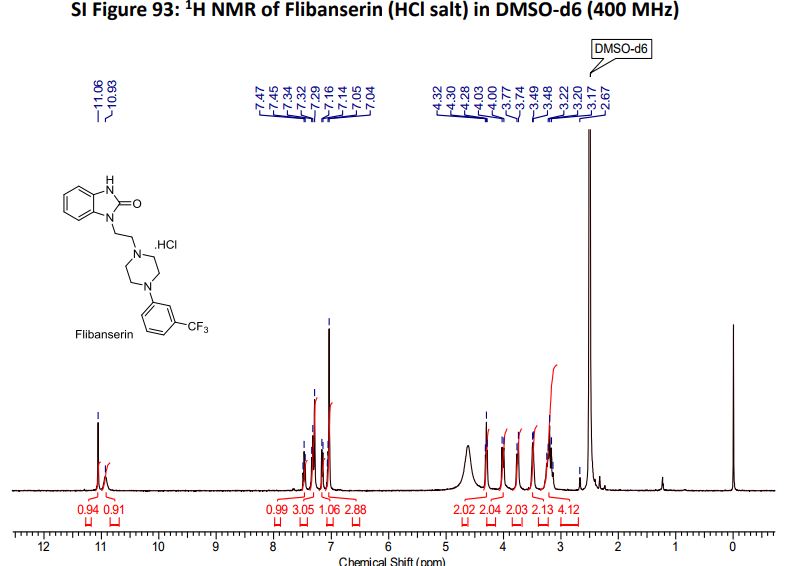
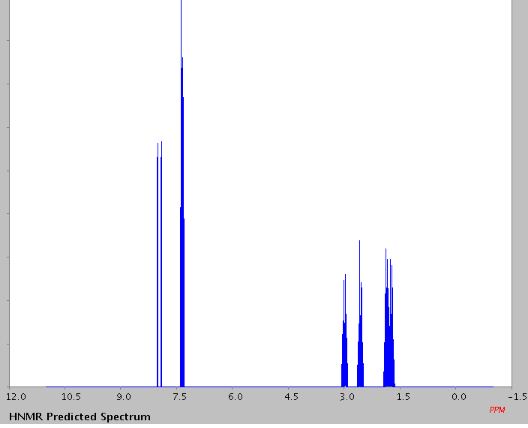
1H NMR (400MHz ,DMSO-d6) 11.06 (s, 1 H), 10.93 (br. s., 1 H), 7.54 – 7.41 (t, J = 7.9 Hz, 1 H), 7.36 – 7.22 (m, 3 H), 7.15 (d, J = 7.6 Hz, 1 H), 7.09 – 7.01 (m, 3 H), 4.30 (t, J = 6.7 Hz, 2 H), 4.01 (d, J = 11.6 Hz, 2 H), 3.75 (d, J = 10.4 Hz, 2 H), 3.54 – 3.43 (d, J = 4.2 Hz 2 H), 3.31 – 3.10 (m, 4 H);
HRMS (ESI): m/z calculated for C20H22ON4F3[M+H]+ 391.1740 found 391.1743;
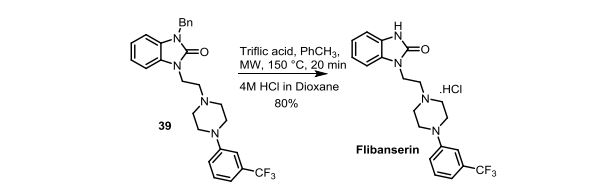

Scheme 4. Synthesis of Flibanserin through Ring Contraction
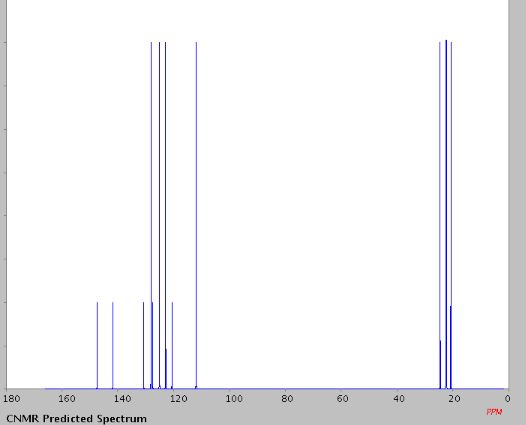
The same methodology was applied for the synthesis of flibanserin, also known as “female viagra”, which is the first approved medication for treating HSDD in women and is classified as a multifunctional serotonin agonist antagonist.(14, 15) Our synthesis of flibanserin commenced with 1-benzyl-1,4-dihydroquinoxaline-2,3-dione 36,(16) which was reacted with known chloride 37(17) under the basic condition in DMF to give the desired product 38 in good yield. Compound 38 was subjected for the decarbonylative cyclization under the optimized condition to afford the product 39 in 59% yield. Finally, the benzyl group was deprotected using trifluoromethanesulfonic acid in toluene under microwave irradiation,(8b, 18) which gave flibanserin in excellent yield (Scheme 4). The final product was isolated as HCl salt, and all of the spectral data are in agreement with the published data.(15c)

Rahul D. Shingare completed his M.Sc (Chemistry) from Fergusson College, Pune in 2008. He worked as a research associate in Ranbaxy and Lupin New drug discovery center, Gurgaon and Pune respectively until 2012 and currently pursuing his doctoral research in NCL – Pune from 2012.
Current Research Interests: Antibacterial Natural Product Hunanamycin A: Total Synthesis, SAR and Related Chemistry.
e-mail: rd.shingare@ncl.res.in

Akshay Kulkarni completed his M.Sc. from Ferguson College, Pune University in the year 2015 and joined our group as a Project Assistant in the month of October, 2015.
Current research interest: Synthesis of silicon incorporated biologically active antimalerial compounds.
e-mail : as.kulkarni@ncl.res.in

Dr.D. Srinivasa Reddy
Organic Chemistry Division
CSIR-National Chemical Laboratory
-
14.
Stahl, S. M. Mechanism of action of Flibanserin, A multifunctional serotonin agonist and antagonist (MSAA), in hypoactive sexual desire disorder CNS Spectrums 2015, 20, 1 DOI: 10.1017/s1092852914000832
-
15.
See, previous synthesis of Flibanserin:
(a) Bietti, G.; Borsini, F.; Turconi, M.; Giraldo, E.; Bignotti, M. For treatment of central nervous system disorders. U.S. Patent 5,576,318, 1996.
(b) Mohan, R. D.; Reddy, P. K.;Reddy, B. V. Process for the preparation of Flibanserin involving novel intermediates. WO2010128516 A2,2010.
(c) Yang, F.; Wu, C.; Li, Z.; Tian, G.; Wu, J.; Zhu, F.; Zhang, J.; He, Y.; Shen, J. A Facile route of synthesis for making Flibanserin Org. Process Res. Dev. 2016, 20, 1576 DOI: 10.1021/acs.oprd.6b00108
-
16.
Jarrar, A. A.; Fataftah, Z. A. Photolysis of some quinoxaline-1,4-dioxides Tetrahedron 1977, 33, 2127 DOI: 10.1016/0040-4020(77)80326-8
-
17.
Xueong, X. Preparation method of Flibanserin. CN104926734 A, 2015.
-
18.
Rombouts, F.; Franken, D.; Martínez-Lamenca, C.; Braeken, M.; Zavattaro, C.; Chen, J.; Trabanco, A. A.Microwave-assisted N-debenzylation of amides with triflic acid Tetrahedron Lett. 2010, 51, 4815 DOI: 10.1016/j.tetlet.2010.07.022
//////////




























 Open Access
Open Access