QbD Validation












QbD Validation Strategies: The Process Design Phase Part 1
Process validation comprises three stages that take place over the life cycle of the pharmaceutical drug product.[1] (See Figure 1.) Stage 1, which will be the focus of this column, is referred to as process design, and encompasses pharmaceutical drug products in early development. Stage 2 is referred to as process qualifications, and includes pharmaceutical drug products at a stage prior to commercialization or prior to submitting a New Drug Application (NDA) or Abbreviated New Drug Application (ANDA). Stage 3 is referred to as continued process verification and includes pharmaceutical drug products that are commercialized and currently marketed products with completed prospective validation. Products that fall in between these categories will need to be assessed on a case-by-case basis and can be placed in the appropriate stage using a “gateway” approach.

Documents for process validation studies at each stage are described in Table I. To proceed from stage 1 to stage 2 (gateway 1), the following documents must be completed and approved internally by the company[2]: a development report, a risk assessment, a control strategy document, and a final manufacturing process description. To proceed from stage 2 to stage 3 (gateway 2) an approved process performance qualification report must be completed and approved as well.
Documents for Process Validation
Table 1
| Stage of Process Validation | Document | Purpose of Document |
| Stage 1
Process Design |
Development report | A detailed history of how product/process was developed including source documents for the control strategy and design space. |
| Risk assessment report | A report conducted early in the development process and updated after completion of development studies and focused on what needs to be studies related to the process. This document is used to develop design space and control strategy. | |
| Control strategy document | This document outlines the control strategy of the process including a list of any CPPs. | |
| Final manufacturing process description document | This document is written based on scale-up experience which describes the intended process for process performance qualification (PPQ). | |
| Stage 2
Process Qualification |
Protocol for process performance qualification (PPD) | Protocol designing the requirement of the process performance quality study |
| PPQ report | Report summarizing the results and outcome of the process performance qualification strategy. | |
| Stage 3
Continued Process Verification (CPV) |
Process risk assessment report | A risk assessment is required for all manufacturing processes. This should be used to determine the frequency of testing defined in the CPV plan. |
| CPV plan | Outlines what should be monitored on an outgoing basis, how the data should be monitored, analyzed, and reviewed. | |
| CPV report | Summaries of the process monitoring data with conclusions and recommendations actions |
Process Design (Stage 1)
Process design is achieved through well-defined and established internal processes that are based on Quality by Design (QbD) guidelines.[3] While QbD is not a mandated FDA requirement, it is recommended to implement a systematic process to evaluate and understand formulation and manufacturing processes using prior knowledge, experimentation and risk assessment. The following describes the key action elements to process design:
1. Develop Quality by Design (QbD) Comprehensive Evaluation: A company should develop consistent internal evaluation systems and identify material attributes (e.g., API and critical excipient) and process parameters, to product critical quality attributes (e.g., CQAs).
2. Identify Critical Quality Attributes (CQAs): All quality attributes for the product should be evaluated by a company based on a quality target product profile (QTPP). Potential CQAs should be identified based on an evaluation of the severity of the attribute in terms of the impact, which is based on clinical experience and in-vitro studies.
3. Provide Risk Assessment: A company must assess the impact of material, process, and environmental variables on the potential CQAs. A risk assessment enables the synthesis of important information for the development of control strategy. This strategy can be revised on an ongoing basis to continue to enhance the company’s understanding of its product and processes.
4. Execute Product/Process Development: A company should conduct process design experiments on generally accepted scientific principles. Only those experiments that result in material being used for clinical trials need to be conducted under cGMP conditions. The subjectivity within a process, such as different API lots, production operators, limitations of commercial manufacturing equipment, environmental conditions, and measurement systems should be considered in the product design process. While validating analytical methods is not necessarily required, analytical methods adopted by the company should be well-defined and provide accurate and consistent results that can be relied upon.
5. Develop Critical Process Parameters (CPPs): CPPs should be developed based on the knowledge gained during process development. CPPs should be consistent with corresponding CQAs.
6. Establish Product/Process Design Space: While not necessary for each operation, design space is developed by relying on knowledge gained from the process development studies and input from is a specific defined process that have been demonstrated to provide quality.
7. Plan Control Strategy: A control strategy is a planned set of controls, derived from product and process understanding which assures process performance and product quality. Control strategies should consider material quality, equipment monitoring and environmental conditions. Control strategies are expected and are needed for stage 2.
In summary, in the process design phase, the process development will develop the manufacturing process. This manufacturing process will yield products with consistent quality that is sufficiently characterized so that the process can be successfully validated to stage 2 process qualification.
[1] The information provided in this article is based upon Guidance for Industry, Process Validation: General Principles and Practices, January 2011, Revision 1.
[2] In general, the development, manufacturing and quality control departments will approve these documents.
[3] See generally, ICH Q8 (R2), Pharmaceutical Development Guidance, November 2009; ICH Q9, Quality Risk Management Guidance, June 2006; ICH Q10, Pharmaceutical Quality System Guidance, April 2009; and US CFR Part 211, Current Good Manufacturing Practice for Finished Pharmaceuticals; 110, 113.
Stages 2 And 3 QbD Validation Strategies For Nonsterile Solid Dosage Forms
Stage 2 is referred process qualification, which includes pharmaceutical drug products at a stage prior to commercialization or prior to submitting a New Drug Application (NDA) or Abbreviated New Drug Application (ANDA). In this stage, it must be demonstrated that the process for manufacture of a drug product is consistent and can produce drug products that are compliant with the Food and Drug Administration’s requirements for filing.
This stage includes two elements. The first element focuses on facility design and equipment installation and maintenance while the second includes process performance qualification (PPQ).[1] Some prerequisites to facility design and equipment installation and maintenance includes validation and qualification of analytical methods, approved standard operating procedures (SOPs) for process validation, implementation of preventive maintenance program (PMs), cleaning validation of equipment, and process specific GMP training. In structuring a PPQ, CPPs and CQAs must be defined, justified, and documented. Other process performance qualification activities must be controlled by an approved protocol that includes the scope, strategy, testing, sampling plan, and acceptance criteria. This study is conducted at a manufacturing site, according to a site validation master plan. Additional strategies are explored below.
Process Qualification Study Strategies: [2]
A strategy needs to be developed for every qualification study and it must be based on deep process understanding gained from the manufacturing experience. Some elements to consider include:
1. Number of Batches: The protocol should include three consecutive batches, with the results summarized in the final process qualification report.
2. Material Selection: Selection of more than one lot of API and critical raw materials should be used during process qualification (especially if the API or critical raw materials are not dissolved or distributed in solution). Based on the selection of these materials, a process should be designed to validate the robustness of the process.
3. Equipment Selection: The process must be qualified on all equipment intended to be used in the manufacture of the product. For equipment determined as being equivalent, qualification on one piece of equipment is sufficient.
4. Design Space/Parameters Ranges: For products that have an established design space, the process qualification should be executed at specified conditions within the design space. For conditions that are high risk, high and low parameter ranges should be considered for the process qualification.
5. Process Re-qualification: Process re-qualification may be required if, for example, a significant deviation from desired process performance is uncovered through stage 3 continued process verification. The process re-qualification studies should bridge back to the original or pivotal clinical biobatch.
Process Qualification Testing:[3]
Process qualification testing should be based on the established CPPs and CQAs and the control strategy and risk assessments, which characterize the product quality and process consistency.
CPP Monitoring: Defined critical process parameters should be monitored and reported in the final qualification report.
- For processes that have a processing fluid (e.g., granulation), microbial testing and hold times will need to be established. For processes where critical ingredients are added as a part of the processing fluid, testing related to the critical ingredient (e.g., assay) at make-up and at the end of hold time should be considered.
- For processes involving lubricated granulation, blend uniformity testing must verify that the active ingredient has been distributed throughout the blended bulk uniformly. To determine unit-dose equivalent (1-3x) blend uniformity of the active ingredient on the final, blend samples should taken from the blender. For combination products, all APIs need to be tested for blend uniformity while respective blenders need to be tested for bilayer tablets.
- For the manufacture of compressed tablets or capsules, during the compression operation a sampling plan should be adopted with around ten evenly spaced intervals throughout the batch processing. After the compression machine is set-up, location one should include the first salable dosage units and location ten should contain the last salable dosage units while location two through nine should be evenly spaced across the lot. For a process using a double sided machine, both the sides of the machine should be sampled at each location. For the manufacture of combination or bilayer tablets, ample samples should be obtained to properly evaluate all API specific tests. Specific to bilayer tablets, samples should be taken to evaluate both layers.
- For the manufacture of film coated tablets, random location sampling should be taken of representative samples. For application of functional membranes, testing should be performed on samples taken from each film coating pan load. For nonfunctional coats, samples may be divided among the film coating pan load.
Acceptance Criteria:
The manufacture process must be validated and reported within the regulatory filing, batch records and final validation report, including all the appropriate specification and procedures (e.g., selected CPPs and release requirements). Any deviation must be investigated and addressed in the validation report.
Each of the processing steps can be analyzed by evaluating the process control charts (based on three sigma limits) and the historical process capability charts. These charts should be evaluated for nonrandom systematic behavior. Ultimately, for all initial process qualification, a comparison must be made to the dissolution profile performed on the biobatch or pivotal reference batch. A batch may be excluded if a nonprocess related assignable cause, like mechanical failure, has been identified.
Statistical Analysis:
Intra-batch and inter-batch variability should be examined from data collected during process qualification through the analysis of process control charts and process capability charts.
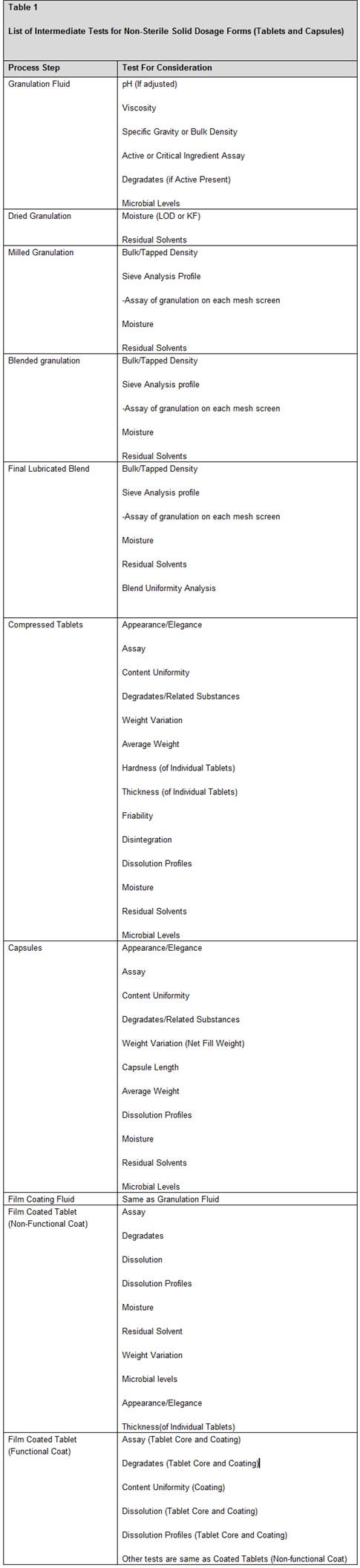
Once a product has completed performance qualification, process validation continues through implementation of continued process verification. This verification includes monitoring operating procedures, preventive maintenance and calibration programs, deviation investigations, annual review, and change control procedures. Any changes to the process must be evaluated through the process change request system and procedures for process change control to determine the impact to on-going process validation. A list of intermediate tests for nonsterile solid dosage forms (tablets and capsules) are compiled in Table 1.
Stage 2 process qualification is conducted at the manufacturing site, which is usually a far distance away from product development/process development facilities. Therefore, another important piece of the process validation includes technology transfer to manufacturing facilities. Technology transfer includes detailed process fit, manufacturing readiness, and an execution phase. Process qualification falls under the execution phase.
Continued Process Verification (Stage 3)[4]
The goal of continued process verification (CPV) is “continual assurance that the process remains in a state of control (validated state) during commercial manufacture.”[5] Once a process has gone through process qualification, an ongoing program to collect and analyze product and process data that relate to product quality is necessary. The objective of the on-going process verification program is to understand the sources of variation, its impact on the process and product attributes, and finally to devise a way to control the variation. The knowledge gained through stage 3 of continued process verification provides ongoing assurance that a product remains in a state of control.
In summary, QbD is not a mandated requirement, however, any pharmaceutical company that instills the QbD approach in their DNA of product development (process design, process qualification, and continued process verification) will come out ahead in their value curve, since we live in an increasingly science-driven regulatory environment in 21st century — where compliance and quality have been essential elements of competitiveness and quality drug products. Hopefully these tips and strategies will support you in designing and validating a quality manufacturing process.
[1] Guidance for Industry, Process Validation: General Principles and Practices at 10.
[2] In drafting this section I relied on the Guidance for Industry, Process Validation: General Principles and Practices, January 2011, at 10-14, as well as my experience in this field.
[3] In drafting this section I relied on the Guidance for Industry, Process Validation: General Principles and Practices, January 2011, ICH Q8 (R2), Pharmaceutical Development Guidance, November 2009; ICH Q9, Quality Risk Management Guidance, June 2006; ICH Q10, Pharmaceutical Quality System Guidance, April 2009; and US CFR Part 211, Current Good Manufacturing Practice for Finished Pharmaceuticals; 110, 113; and my experience in this field.
[4] In drafting this section I relied on the Guidance for Industry, Process Validation: General Principles and Practices, January 2011, at 14-16 as well as my experience in this field.
[5] Guidance for Industry, Process Validation: General Principles and Practices, January 2011, at 14.
24,510 total views, 2 views today
