QbD IN PHARMACEUTICAL INDUSTRY













INTRODUCTION1-5
“Quality by design means designing and developing manufacturing processes during the product development stage to consistently ensure a predefined quality at the end of the manufacturing process.”

Fig.1: Quality by Design2
The concept of QbD was mentioned in the ICH Q8guideline, which states that “quality cannot be tested into products i.e., quality should be built in by design.” According to ICH Q8 QbD is defined as a systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management.
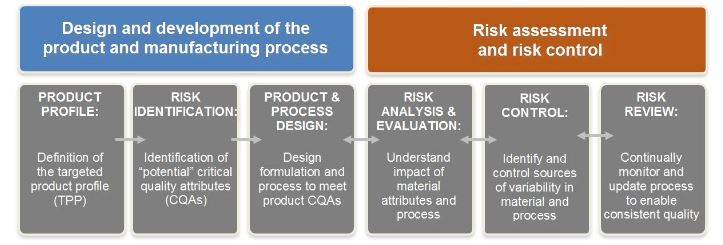
Quality by Design (QbD) has become a new concept for development of quality pharmaceutical products, It is an essential part of the modern approach to pharmaceutical quality, QbD is a best solution to build a quality in all pharmaceutical products but it is also a major challenge to the Pharmaceutical industry whose processes are fixed in time, despite inherent process and material variability, Under this concept of QbD throughout designing and development of a product, it is essential to define desire product performance profile [Target product Profile (TPP), Target Product Quality Profile (TPQP)] and identify critical quality attributed (CQA). On the basis of this we can design the product formulation and process to meet the product attributes. This leads to recognize the impact of raw materials [critical material attributes (CMA), critical process parameters (CPP) on the CQAs and identification and control sources of variability. QbD is an emerging idea which offers pharmaceutical manufacturer with increased self-regulated flexibility while maintaining tight quality standards and real time release of the drug product.
ICH 2,3,6,7
ICH Q8, Q9, Q10 & Q11are designed as separate but linked in a series of documents exploring pharmaceutical products lifecycle
ICH Q8 Pharmaceutical Development
ICH Q9 Quality Risk Management
ICH Q10 Pharmaceutical Quality System
ICH Q11 – Development and Manufacture of Drug Substance.

Fig.2: Concept of QbD3
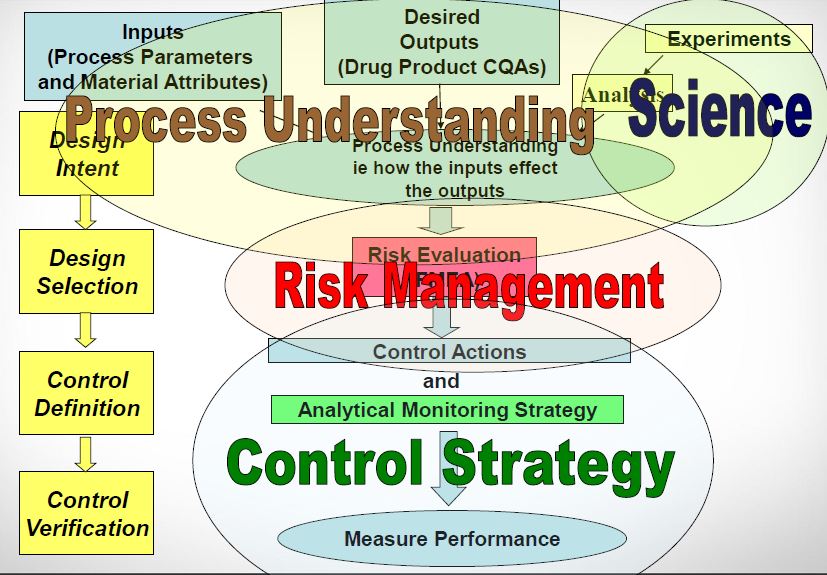
Quality by design system

Fig.3: QbD system7
Steps 8,9,10
a. Development of new molecular entity
· Preclinical study.
· Nonclinical study.
· Clinical Study.
· Scale up.
· Submission for market Approval
b. Manufacturing
· Design Space.
· Process Analytical Technology.
· Real time Quality Control.
c. Control strategy
· Risk based decision.
· Continuous Improvement.
· Product performance
Benefits of QbD 1,3,5,6
1. QbD is good Business.
2. Eliminate batch failures.
3. Minimize deviations and costly investigations.
4. Avoid regulatory compliance problems.
5. Organizational learning is an investment in the future.
6. QbD is good Science.
7. Better development decisions.
8. Empowerment of technical staff.
Opportunities 8,9
1. Efficient, agile, flexible system.
2. Increase manufacturing efficiency; reduce costs and project rejections andv waste.
3. Build scientific knowledge base for all products.
4. Better interact with industry on science issues.
5. Ensure consistent information
6. Incorporate risk management.
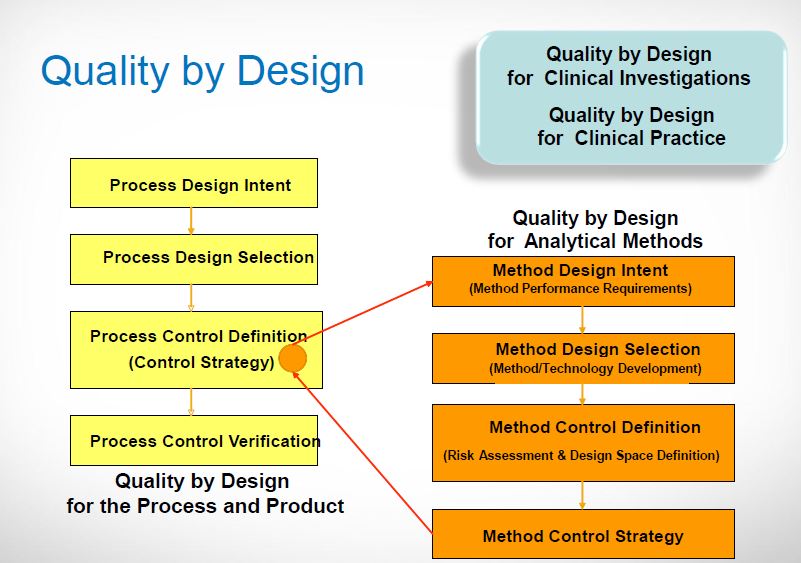
Process [11, 12, 13]
Quality by Design is a scientific risk based holistic and proactive approach to pharmaceutical development. It involves the designing and planning of a drug product and process before actual experiment. Over the past several years, pharmaceutical scientists have provided several more specific definitions of the elements of quality by design and a draft of an annexure to ICH Q8 has been released.

Fig.4: Process[11]
Elements
· Identifying Target Product Quality Profile (TPQP).
· Identifying CQA’s.
· Design product and defining product design space.
· Process design and defining process design space.
· Defining control strategy.
· Process validation.
· Regulatory filings.
· Process monitoring, life cycle management and continuous improvement.
Quality by Design Tools
Target product profile(TPP).14, 15
The role of target product profile (TPP) is to serve as a tool for “quality planning” for the drug product with “the end in mind” i.e. a summary of the drug development program described in the context of prescribing information goals. A quality target product profile (QTPP) is a term which is a natural extension of TPP for product quality .A QTPP relates to the quality of a drug substance or the drugs products that is necessary to deliver a desired therapeutic effect .QTPP is a predetermined summary of the characteristics of the drug product that will ideally be essential to ensure the desired quality with respect to safety and efficacy of the product. These predetermined QTPP evolve over time during drug development and may be modified to incorporate new knowledge, as is warranted by ongoing clinical studies such a dose effect and toxicology data.
Critical quality attributes (CQA).
Critical quality attributes are defined as physical, chemical, biological or microbiological properties or characteristics that need to be controlled to ensure product quality.( According to ICH Q8) CQAs as physical, chemical, biological or microbiological properties or characteristics that should be within an appropriate limit, range, or distribution to ensure the desired product quality. CQA has been used by some to describe elements of the TPQP while others have used CQA to describe mechanistic factors that determine product performance. Thus CQA is used to describe both aspects of product performance and determinants of product performance.
Risk assessment16, 17, 18
A key objective of risk assessment in pharmaceutical development is to identify which material attributes and process parameters affect the drug product CQAs, that is, to understand and predict sources of variability in the manufacturing process so that an appropriate control strategy can be implemented to ensure that the CQAs are within the desired requirements.
The identification of critical process parameters (CPP) and critical material attributes is an iterative process and occurs throughout development. During the initial phases of development, prior knowledge serves as the primary basis for the designation as there is not sufficient process/product understanding on the product under development. Therefore, the risks identified at the initial phases are perceived risks and as further process/product understanding is gained, the actual risks become clearer and a control strategy can be better defined. The risk assessment tools used in earlier phases of development therefore tend to be more qualitative and serve as a means to prioritize the experimentation.
Design space
ICH Q8 defines design space as, the multidimensional combination and interaction of input variables (material attributes) and process parameters that have been demonstrated to provide assurance of quality. Moving out of the design space is considered to be a change and would normally initiate a regulatory post-approval change process. The design space is proposed by the applicant and is subject to regulatory assessment and approval. Design space is potentially scale and equipment dependent, the design space determined on the laboratory scale may not be relevant to the process at the commercial scale. Therefore, design-space verification at the commercial scale becomes essential unless it is confirmed that the design space is scale-independent. Currently, generic drug sponsors obtain information about acceptable ranges for individual CPPs and CMAs at laboratory or pilot scales.
Multidimensional combination of and interaction of input variables and process parameters that have been demonstrated to provide Quality Assurance

Fig.5: Design space17
Control Strategy19, 20
A planned set of controls, derived from current product and process understanding that ensures process performance and product quality…..” ICH Q8 (R2) & Q10
Control Strategy includes following elements (but not limited to):
· Input material attributes (e.g. drug substance, excipients, container closure).
· Equipment operating conditions (process parameters).
· In-process controls.
· Finished product specifications.
· Controls for each unit operations.
· Methods and frequency of monitoring and control.
Life cycle Management and Continuous improvement20-23
After approval, CQAs are monitored to ensure that the process is performing within the defined acceptable variability that served as the basis for the filed process design space. The primary benefit of an expanded process design space would be a more flexible approach by regulatory agencies. In the QbD paradigm, process changes within the design space will not require review or approval. Therefore, process improvements during the product life cycle with regard to process consistency and throughput could take place with fewer post approval submissions. In addition to regulatory flexibility, the enhanced understanding of the manufacturing process would allow more informed risk assessment as per ICH Q9 regarding the affects of process changes and manufacturing deviations on product quality. Manufacturing, the experience grows and opportunities for process improvement identified are operating space could be revised within the design space without the need for post-approval submission. Over the lifetime of a product, process changes may be required to be made and may require process characterization, validation and filing of the changes to the approved process design space. The quality system needs to provide adequate oversight during QbD implementation of changes that will not go through regulatory approval. Robustness of the quality system would need to be demonstrated with respect to the following four elements: process performance/product quality monitoring; preventative/corrective action, change management and management review of process performance and product quality
Problems in implementing QbD
1. Internal misalignment.
2. Lack of belief in business case.
3. Lack of technology to execute.
4. Alignment with third parties.
5. Inconsistency of treatment of QbD across FDA.
6. Lack of tangible guidance for industry.
7. Regulators not prepared to handle QbD application.
8. The way promised regulatory benefits are currently being shared dose not inspire confidence.
9. Misalignment of international regulatory bodies.
10. Current interaction with companies is not conducive to QbD.
CONCLUSION
The goal of QbD is to reduce product variability and defects, thereby enhancing patient efficacy and safety. Furthermore, it also helps in enhancing the product and process development depth and understanding which then directly improves the efficiency and helps to effectively manage the post approval changes. The elements QbD-QTPP, CQA, CMA and CPP enhance the product development process leading to a thorough product and process understanding, successful scale-up, control strategy and continual improvement. Finally, product and process capability is continually reviewed and improved post approval during product lifecycle management.
REFERENCES
1. Woodcock J, The concept of pharmaceutical quality. American Pharmaceutical Review, 7(6), 2004, p.10–15.
2. Q9: Quality Risk Management. ICH Harmonized Tripartite Guidelines International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2006.
3. Q10: Pharmaceutical Quality System, ICH Tripartite Guidelines. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2007.
4. Lionberger RA, Lee LS, Lee L, Raw A ,Yu LX, Quality by design: Concepts for ANDAs, The AAPS Journal, 10, 2008, p.268–276.
5. FDA Guidance for Industry and Review Staff: Target Product Profile – A Strategic Development Process Tool (Draft Guidance).
6. Q8 (R1): Pharmaceutical Development, Revision 1, ICH Harmonized Tripartite Guidelines, International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2007.
7. Food and Drug Administration, Office of Generic Drugs White Paper on Question-based Review: fda.gov/cder/ OGD/QbR.htm.
8. Callis JB, Illman DL, Kowalski BR, Process analytical chemistry. Analytical Chemistry, 59, 1987, 624A–637A.
9. Yu LX, Pharmaceutical quality by design: Product and process development, understanding, and control. Pharmaceutical Research, 25, 2008, 781–791.
10. Munson J, Gujral B, Stanfield CF, A review of process analytical technology (PAT) in the U.S. pharmaceutical industry. Current Pharmaceutical Analysis, 2, 2006, 405–414.
11. Juran JM. On quality by design. The new steps for planning quality into goods and services New York free press 1992 p.no.1-2.
12. MN Nasr. Implementation of quality by design (QbD): status, challenges, and next steps. FDA Advisory Committee for Pharmaceutical Science. Available at: fda.gov/ohrms/dockets/ac/06/slides/2006-4241s1_6.ppt (accessed 9/7/2015).
13. ISPE PQLI. Draft PQLI summary update report. . Org/cs/pqli_product_quality_lifeycle_implementation_/draft_pqli_summary_Update- report (accessed 11/21/2007).
14. Food and Drug Administration: fda.gov/ohrms/ dockets/ac/06/minutes/2006–4228m1.pdf, 2006.
15. US Food and Drug Administration, Guidance for Industry: Q10 Pharmaceutical Quality Systems, 2009.
16. Guidance for industry “Q9 Quality Risk Management” by US department of Health and Human Services, Food and drug Administration, Center for Drug and Evaluation Research, June 2006.
17. WHO Guideline on Quality Risk Management A draft guidance August 2010.
18. Implementation of ICH Q9 in the pharmaceutical field an example of methodology from PIC/S 2010.
19. Gibson, M, Product Optimization: Pharmaceutical Preformulation and Formulation. Taylor & Francis, New York 2001.
20. Seely, RJ, Haury, J, in: Rathore, AS, Sofer, G (Eds.), Process Validation in Manufacturing of Biopharmaceuticals, Taylor & Francis, Boca Raton, FL 2005, pp.13-50 .
21. US Food and Drug Administration. guidance for industry: Q10 quality systems approach to pharmaceutical cGMP regulations (FDA, Rockville, MD, September 2006)
22. US Food and Drug Administration. Pharmaceutical cGMPs for the 21st century: a risk-based approach (FDA, Rockville, MD, August 2002). fda. gov/oc/guidance/gmp.html.
23. Rathore KS, Pokhrana D, Yadav S, Quality by Design, articlesbase.com/college-and-university-articles/quality-by-design-qbd-4890152.html (accessed on July4, 2015).

FDA
INTRODUCTION[1-5]
The FDA publication defined Quality by Design as:
- Developing a product to meet predefined product quality, safety and efficacy
- Designing a manufacturing process to meet predefined product quality, safety and efficacy
The use of QbD principles during product development provides opportunities to facilitateinnovation and continual improvement throughout the product lifecycle, compared to traditionalapproaches hence it is systematic way to product and process development. QbD principles increase process knowledge and product understanding, often through the application of new technologies such as PAT or modeling. The increased process knowledge and product understanding resulting from QbD can increase the efficiency of manufacturing processes; reduce product recalls and compliance actions, resulting in cost savings for pharmaceutical companies. By reducing uncertainty and risk, QbD can allow industry and regulators to focus their resources in the most critical areas. Because much more process understanding has been demonstrated and expressed in the dossier, QbD filings also can help facilitate CMC reviews and GMP inspections by the regulators and decrease the number of post-approval regulatory submissions required to make process changes. QbD can also facilitate the use of innovative technologies and promote the use of new approaches to perform process validation, such as continuous quality verification.
NEED OF QBD[6]
- Higher level of assurance of product quality
- Cost saving and efficiency for industry and regulators
- Facilitate innovation to address unmet medical needs
- Increase efficiency of manufacturing process and reduce manufacturing cost and product rejects
- Minimize/eliminate potential compliance actions, costly penalties and recalls
- Enhance opportunities for first cycle approval
- Streamline post approval manufacturing changes and regulatory processes
- More focused PAI and post approval cGMP inspections
- Opportunities for continual improvement
The main objectives of Quality by Design
- To facilitate innovation and continuous improvement throughout the product lifecycle
- To provide regulatory flexibility for specification setting and post-approval changes
- To streamline the submission and review processes
ELEMENT OF THE QBD
1.Define Target Product Quality Profile
2.Identify Critical Quality Attributes, Process Parameters, and Sources of Variability
3.Design Product and Manufacturing Processes
4.Control Manufacturing Processes to Produce Consistent Quality over Time
5.Risk assessment & risk control
6.Process Design and Development
Define Target Product Quality Profile (TPQP)
The target product profile (TPP) has been defined as a “prospective and dynamic summary of the quality characteristics of a drug product that ideally will be achieved to ensure that the desired quality, and thus the safety and efficacy, of a drug product is realized”. This includes dosage form and route of administration, dosage form strength(s), therapeutic moiety release or delivery and pharmacokinetic characteristics (e.g., dissolution and Excipients meeting specification Unit operation with fixed process parameters In process specification Finished product aerodynamic performance) appropriate to the drug product dosage form being developed and drug product quality criteria (e.g., sterility and purity) appropriate for the intended marketed product .The Target Product Quality Profile (TPQP) is a term that is a natural extension of TPP for product quality. It is the quality characteristics that the drug product should possess in order to reproducibly deliver the therapeutic benefit promised in the label. The TPQP guides formulation scientists to establish formulation strategies and keep formulation efforts focused and efficient.TPQP is related to identity, assay, dosage form, purity, stability in the label .
Identify Critical Quality Attributes, Process Parameters, And Sources Of Variability:
A pharmaceutical manufacturing process is usually comprised of a series of unit operations to produce the desired product. A unit operation is a discrete activity that involves physical changes, such as mixing, milling, granulation, drying, compaction, and coating A physical, chemical or microbiological property or characteristic of an input or output material is defined as an attribute. Process parameters include the type of equipment and equipment settings, batch size, operating conditions (e.g., time, temperature, pressure, pH, and speed), and environmental conditions such as moisture The quality and quantity of drug substance and excipients are considered as attributes of raw materials. To demonstrate the reproducibility and consistency of a process, process capability should be studied. Process capability is a statistical measure of the inherent process variability for a given characteristic. The most widely accepted formula for process capability is six sigma. Process capability index19 is the value of the tolerance specified for a particular characteristic divided by the process capability, which is defined as follows
Upper limit of specification –lower limit of specification
Process capability index (CpK) = ———————————-
standard deviation
If the CpK value is significantly greater than one, the process is deemed capable. If the process capability is low, Strong recommends an iterative five step procedure to progressively reduce the variability of the process.
These five steps are:
1. Define: The intended improvement should be clearly stated. This may be defined as material attributes. Should be measured to see if they are out of specification the out of specification data should be analyzed and used to the sigma level of the process.
2. Measure: The critical product performance attributes
3. Analyze: When the sigma level is below the target, steps should be taken to increase it, starting by identifying the most significant causes of the excessive variability.
4.Improve: The process should be redesigned and/or process controls should be incorporated to eliminate or attenuate the significant root causes of variance.
5. Control: The improved manufacturing process should be evaluated and maintained.
Design Product and Manufacturing Process
In order to design and develop a robust generic product that has the desirable TPQP, a product development scientist must give serious consideration to the biopharmaceutical properties of the drug substance. These biopharmaceutical properties include physical, chemical, and biological properties. Physical properties include physical description (particle size, shape, and distribution), polymorphism, aqueous solubility as function of pH, hygroscopicity, and melting points. Pharmaceutical solid polymorphism, for example, has received much attention recently. Its impact on product quality and performance has been discussed in recent review articles. Chemical properties include pKa, chemical stability in solid state and in solution as well as photolytic and oxidative stability while biological properties include partition coefficient, membrane permeability, and/or oral bioavailability. Biopharmaceutical properties should be assessed for every form for which there is an interest in development and every form that can potentially be created during processing (e.g., hydrates, anhydrates) or in vivo (e.g., less soluble salts, polymorphic forms, hydrates). The investigation of these properties is termed preformulation in pharmaceutical science.
The goal of preformulation studies is to determine the appropriate salt and polymorphic form of drug substance evaluate and understand its critical properties, and generate a thorough understanding of the material’s stability under various processing and in vivo conditions, leading to an optimal drug delivery system. Pharmaceutical preformulation studies need to be conducted routinely to appropriately align dosage form components and processing with drug substance and performance criteria. Mechanical properties, though not often studied in detail, can have a profound impact on solid dosage form development and processing. A sound understanding of mechanical properties of the drug and excipients can be useful in developing a processing method such as granulation or direct compression, rationally selecting excipients whose properties can compensate for the properties of the drug substance, and helping assess critical material attributes and root cause analysis during process scale-up or failure. Pharmaceutical materials can be elastic, plastic, viscoelastic, hard, soft, tough, or brittle. There exist various methods in the literature to evaluate these mechanical properties. The knowledge of mechanical properties of the drug and excipients are expected to play a more significant role in product design and development in the future. Drug-excipient compatibility has been identified as one of the most frustrating, troubling, and perplexing formulation challenges. Despite the fact that excipients can alter stability and bioavailability of drugs, the general principles of selecting suitable excipients for dosage forms are not well defined, and excipients are often selected without systematic drug-excipient compatibility testing. To avoid costly material wastage and time delays, ICH Q8 recommends drug-excipient compatibility studies to gain early prediction of drug-excipient compatibility. Systematic drug-excipient compatibility studies offer several advantages: minimizing unexpected stability problems which usually lead to increases in time and cost; maximizing the stability of a formulation; and enhancing understanding of drug-excipient interactions that can help with root cause analysis if stability problems occur. Despite its significance, however, there is no universally accepted way to conduct drug-excipient compatibility studies in this evolving area. One method is thermal analysis, where a physical property of a substance (e.g., melting point) and/or reaction products is measured as a function of temperature while the substance is subject to a controlled temperature program. Another method utilizes isothermal stress. This method typically involves storing the drug-excipient blends or compacts with or without moisture at elevated temperature and determining drug content or degradation product formation as a function of time. Both methods can be used together to evaluate the compatibility of drugs with the selected excipients.
Control Manufacturing Processes To Produce Consistent Quality Over Time
This means that manufacturers have knowledge of the operating range as well as the proven range of critical raw material attributes and process parameters. The operating range is defined as the upper and/or lower limits for raw material attributes and process parameter values between which the attribute and parameter are routinely controlled during production in order to assure reproducibility. The proven range 20 is defined as the upper and/or lower limits for process parameter values between which the parameter is known to produce a high quality product that delivers the therapeutic benefit claimed on the label. The proven range can be established based on historical and/or experimental data. It can also be established based on scientific and operational judgment and expertise .Within the QbD, design space21 is defined as then multidimensional combination and interaction of input variables (e.g., material attributes) and process parameters that have been demonstrated to provide quality assurance
Risk Assessments
Several applications in the CMC pilot included risk assessments, especially for the drug product by linking input and process variables to CQAs. Tools used in the risk assessment included the Ishikawa or fishbone diagram, failure mode effect analysis (FMEA), and Pareto analysis. An Ishikawa or fishbone diagram is used to identify all potential variables, such as raw materials, compression parameters, and environmental factors, which can have an impact on a particular CQA, such as tablet hardness. A FMEA can then be used to rank the variables based on risk (i.e., a combination of probability, severity, and detectability) and to select the process parameters with higher risks for further studies to gain greater understanding of their effects on CQAs. A multidisciplinary team based on prior knowledge and experiments amasses the risk assessment. “It isimportant to provide a systematic risk analysis of how raw materials, process steps, and process parameters affect product quality,” One of the points to consider in risk assessment, is to provide an explanation when citing prior experience as the basis for assigning risk. The risk assessment that leads to the development of a comprehensive control strategy to reduce risk to product quality should be described, and the risk reduction and control should be discussed for changes that occur inside or outside the design space, “Risk assessment can provide increased assurance to quality,” because “process variability is identified and its linkage to product CQAs is understood; process and product controls reduce the impact of variability; and quality product will continue to be made when movement within the design space occurs in the future.” A risk assessment also is important for effective communication between FDA and industry and for intra company communication (such as between research/development and manufacturing and among multiple manufacturing sites), “And within FDA, risk assessment allows for a dialogue between pre-and post-marketing review functions and among review, compliance, and field inspection staffs.
Process Design And Development
Strictly speaking, process and product design and development cannot be separated since a formulation cannot become a product without a process. Process design is the initial stage of process development where an outline of the commercial manufacturing processes is identified on paper, including the intended scales of manufacturing. This should include all the factors that need to be considered for the design 22 of the process, including facility, equipment, material transfer, and manufacturing variables other factors to consider for process design are the target product quality profiles. Depending upon the product being developed, type of process, and process knowledge the development scientists have, it may be necessary to conduct preliminary feasibility studies before completing the process design and development. The selection of type of process depends upon the product design and the properties of the materials.
CONCLUSION
Ensures robust commercial manufacturing methods for consistent production of quality drugs.reduce product recalls and compliance actions, resulting in cost savings for pharmaceutical companies. Ensures the consumers that therapeutic equivalent generics are manufactured every single time. Offers the agency that quality applications are submitted to improve the review efficiency and to reduce the application approval times. QbD methodology helps in identifying and justifying target product profiles, product and process understanding.Helps in continuous improvement. There is a need for vigorous and well funded research programs to develop new pharmaceutical manufacturing platforms.
REFERENCE
1. FDA Guidance for Industry –Analytical Procedures and Method Validation, Chemistry, Manufacturing, and Controls Documentation, Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER), August 2000.
2. International Conference on Harmonization Quality Guidelines Q2(R1), Validation of Analytical Procedures, Text and Methodology, Parent guideline dated 27 Oct 1994, Complementary guideline on methodology dated 6 Nov 1996, incorporated November 2005.
3. US Food and Drug Administration. Guidance for Industry. PAT—A Framework for Innovative Pharmaceutical Manufacturing and Quality Assurance. Pharmaceutical cGMPs. Rockville, MD, 2004 Sept., 1–21.
4. ICH Q8 (R1), 2008b. Pharmaceutical Development. Part I: Pharmaceutical Development, a. And Part II: Annex to Pharmaceutical Development,MEDIA4986.pdf.
5. “Quality by Design (QbD) – A Modern System Approach to Pharmaceutical Development and Manufacturing – FDA Perspective” Moheb M. Nasr
6. A New Pharmaceutical Quality Assessment System (PQAS) for the 21st Century, AAPS Workshop October 2010
7. Woodcock J. Pharmaceutical quality in the 21st Century—an integrated systems approach, December 16, 2009.
8. Joshi Y, LoBrutto R, Serajuddins ATM. Industry opinion of regulatory influence: the initiative on pharmaceutical cGMPs for the 21st century. Journal of Process Analytical Technology July/August 2010.
9. Nasr MM. A New Pharmaceutical Quality Assessment System (PQAS) for the 21st Century, AAPS Workshop October 2010.
10. QbD Principles to be implemented in future FDA guidance; DIA Dispatch, Oct. 28, 2010.
11. Yu, LX. Implementation of quality by design: question Based Review, DIA Meeting, June 2010.
12. Chen C. Implementation of ICH Q8 and QbD—an FDA Perspective, Pharma Forum Yokohama.
ABBREVIATIONS
Qbd Quality By Design
FDA Food and Drug Administration
NDA New Drug Application
PAT Process Analytical Technology
cGMP Current Good Manufacturing Practices
ICH International Conference On Harmonization
CQAS Critical Quality Attributes
CPPS Critical Process Parameters
FMEA Failure Mode and Effects Analysis
TPQP Identify Target Product Quality Profile TPQP
API Active Pharmaceutical Ingradient
INTRODUCTION
Quality by design (QbD) encompasses designing and developing formulations and manufacturing processes which ensures predefined product specifications. In 2002, the FDA announced a new initiative (cGMP for the 21st Century: A Risk based Approach). ICH Q8 defines quality as “The suitability of a drug either substance or drug product for its intended use. This term includes such attributes as the identity, strength, and purity. “Quality by Design” A systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management” From guidance to Pharmaceutical Development. ICH Q8 guideline (International Conference for the Harmonization of pharmaceutical regulation). Quality by Design is a systematic scientific approach to development and design of products and processes illustrated and facilitated through the establishment of the Design Space. The FDA’s Janet Woodcock has frequently stated that QbD is derived from a combination of prior knowledge, experimental assessment, and a cause-and-effect model that links critical process parameters and critical quality attributes. Achieving the goal of manufacturing process excellence through QbD requires us to begin the work in process development. The FDA’s Process Analytical Technology (PAT) guideline shows the value of continuous learning that comes from analyzing process data when coupled with systems that support the acquisition of knowledge from those data, saying: “Continuous learning through data collection and analysis over the lifecycle of a product is important. These data can contribute to justifying proposals for post-approval changes. Approaches and information technology systems that support knowledge acquisition from such databases are valuable for the manufacturers and can also facilitate scientific communication with the Agency”.
BENEFITS OF QBD [6]
- Better understanding of the process
- Less batch failure.
- Reduction of post-approval submissions.
- Better innovation due to the ability to improve processes without resubmission to the FDA when remaining in the Design Space.
- More efficient technology transfer to manufacturing.
- Greater regulator confidence of robust products.
- Innovative process validation approaches
- Improved yields, lower cost, less investigations, reduced testing, etc.
- Continuous improvement over the total product life cycle (i.e. controlled, patient guided variability).
- Improved yields, lower cost, less investigations, reduced testing, etc.
QBD DEVELOPMENT PROCESS MAY INCLUDE
a. Begin with a target product profile that describes the use, safety and efficacy of the product
b. Define a target product quality profile that will be used by formulators and process engineers as a quantitative surrogate for aspects of clinical safety and efficacy during product development
c. Gather relevant prior knowledge about the drug substance, potential excipients and process operations into a knowledge space. Use risk assessment to prioritize knowledge gaps for further investigation
d. Design a formulation and identify the critical material (quality) attributes of the final product that must be controlled to meet the target product quality profile
e. Design a manufacturing process to produce a final product having these critical materials attributes.
f. Identify the critical process parameters and input (raw) material attributes that must be controlled to achieve these critical material attributes of the final product. Use risk assessment to prioritize process parameters and material attributes for experimental verification. Combine prior knowledge with experiments to establish a design space or other representation of process understanding.
g. Establish a control strategy for the entire process that may include input material controls, process controls and monitors, design spaces around individual or multiple unit operations, and/or final product tests. The control strategy should encompass expected changes in scale and can be guided by a risk assessment.
h. Continually monitor and update the process to assure consistent quality
TRADITIONAL APPROACH & ENHANCED QBD APPROACH [6]
|
Aspect |
Traditional approach |
QbD approach |
|
Pharmaceutical Development |
Empirical, Random, Focus on optimization |
Systematic, Multivariate experiments, Focus on control strategy and robustness |
|
Manufacturing Process |
Fixed |
Adjustable within design space, managed by company’s quality systems |
|
Process Control |
Some in-process testing |
PAT utilized, Process operations tracked and trended |
|
Product Specification |
Primary means of quality control, based on batch data |
Part of the overall quality control strategy, based on desired product performance |
|
Control Strategy |
By testing and inspection |
Risk-based control strategy , real-time release possible |
BUILDING BLOCKS OF QBD[7]
The first step to implement Quality by Design is to understand critical output of QbD and after that identify critical building blocks of QbD such as improving process understanding and risk associated with it.
- Critical Quality Attributes (CQAs):The critical process output measurements linked to patient needs .
- Critical Process Parameters (CPPs):The process inputs (API, Excipients), process control and environmental factors that have major effects on the Critical Quality Attributes (CQAs) .
- Design Space:The combination of input variables and process parameters that provide quality assurance [3].
- Failure Mode and Effects Analysis (FMEA):It examines raw material variables, Identifies how a process can fail and the areas of process that remains at greatest risk of failing .
- Process model:A quantitative picture of process based on fundamental and statistical relationship that predict the critical quality attribute (CQA) result .
- Process Capability:It tracks process performance relative to CQA specification and provide measurement repeatability and reproducibility regarding CQAs .
- Process Robustness:The ability of process to perform when faced with uncontrolled variation in process, input and environmental variables .
- Process Control:It is a control procedures including statistical process control that keeps the processes and measurement system on target and within desired variations .
- Raw material factors:It includes the stability and capability of raw material manufacturing processes that affect process robustness, process capability and process stability .
- Risk level:It is a function of the design space, FMEA result and process and measurement capability, control and robustness .
STEPS –FOR QUALITY BY DESIGN APPROACH[8-12]
Step 1. Idenify the Target Product Profile Profile
Step 2. Critical Quality Attributes (CQAS)
Step 3. Link the drug and excipient attributes and the process parameters to the CQAS
Step 4. Define the Design Space
Step 5. Define the Control Strategy
Step 6. Product lifecycle management & continual improvement
Step1 : Identify Target Product Quality Profile (TPQP)
The target product profile (TPP) has been defined as a “prospective and dynamic summary of the quality characteristics of a drug product that ideally will be achieved to ensure that the desired quality, and thus the safety and efficacy, of a drug product is realized”. This includes dosage form and route of administration, dosage form strength(s), therapeutic moiety release or delivery and pharmacokinetic characteristics (e.g., dissolution and Excipients meeting specification Unit operation with fixed process parameters In process specification Finished product aerodynamic performance) appropriate to the drug product dosage form being developed and drug product-quality criteria (e.g., sterility and purity) appropriate for the intended marketed product .The Target Product Quality Profile (TPQP) is a term that is a natural extension of TPP for product quality. It is the quality characteristics that the drug product should possess in order to reproducibly deliver the therapeutic benefit promised in the label. The TPQP guides formulation scientists to establish formulation strategies and keep formulation efforts focused and efficient. TPQP is related to identity, assay, dosage form, purity, stability in the label .
Step 2: Identifying CQAs
Once TPP has been identified, the next step is to identify the relevant CQAs. A CQA has been defined as “a physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality”. Identification of CQAs is done through risk assessment as per the ICH guidance Q9 Prior product knowledge, such as the accumulated laboratory, nonclinical and clinical experience with a specific product-quality attribute, is the key in making these risk assessments. Such knowledge may also include relevant data from similar molecules and data from literature references. Taken together, this information provides a rationale for relating the CQA to product safety and efficacy.
Step 3: Link the drug and excipient attributes and the process parameters to the CQAs
Inputs to the process control variability of the Output. Y= f(X) Where, X – Inputs i.e. process parameters (people, Equipment, Measurement, process, material, and environment, Y- Quality attributes is the output. Design of experiments is an efficient method to determine relevant parameters & interactions. The experimental approach for identifying parameters can includes the following methods
(1) To choose experimental design,
(2) Conduct randomized experiments
(3) To analyze data
To Link the drug and excipient attributes and the process parameters to the CQAs there includes a formal risk management process.
Step 4: Design Space
The multidimensional combination and interaction of input variables (e.g., material attributes) and process parameters that have been demonstrated to provide assurance of quality. Working within the design space is not considered as a change. Movement out of the design space is considered to be a change and would normally initiate a regulatory post approval change process. Design space is proposed by the applicant and is subject to regulatory assessment and approval. It’s a Key for claiming Process understanding (pharmaqbd.com, 2011), which establishes a link between the attributes of the drug product and process parameters, process attributes and material attributes of the active pharmaceutical ingredient (API) and excipients that go into the drug product. From a practical standpoint, process understanding, and the associated design space, entails: identifying and explaining all critical sources of variability; managing variability by the process via measurement and control of critical process variables; and reliably and accurately predicting and controlling product attributes within specifications (achieve quality). Process understanding is Key for Quality Risk Management & QRM is the base for any Control Strategy.
Step: 5 Define the Control Strategy
Justification of necessary controls of input material attributes (e.g., drug substance, adhesive polymer, primary packaging materials) based on an understanding of their impact on process ability or product quality In-Process Controls & End Product Controls (if necessary).It is based on Process and Formulation Understanding that Drives the Process in the Design Space Based on Quality Risk Management to ensure conforming Quality according Specifications.
Step: 6 Product lifecycle management and continual improvement
Use accumulating manufacturing data as the basis to modify and improve the process within the design space. At this stage, the underlying concepts and rationale for implementing quality-by-design practices are well understood and accepted. Despite this progress, there remain critical impediments to QbD implementation including the following:
From a practical standpoint, what comprises and how does one acquire process understanding? From a practical standpoint, how does one decide that a process parameter is critical? And most importantly, how do we know that the identified design space of the process links to the clinical design space of the patient? After all, the aim is to design a process that meets the needs of safety and efficacy for the patient.
CONCLUSION
Quality by design is an essential part of the modern approach to pharmaceutical quality. QbD is novel approach which is currently being used in pharmaceutical industry than empirical approaches of the product development because it reduces the product variability.A prospective and dynamic summary of the quality characteristics of a drug product that ideally will be achieved to ensure that the desired quality, and hence the safety and efficacy, of a drug product is realized.
REFERANCES
1. Food and Drug Administration. Final Report on Pharmaceutical cGMPs for the 21st Century – A Risk Based Approach, fda.gov/ cder/ gmp/ gmp 2004/ GMP_ final report 2004
2. ICH Q8 — Guidance for Industry, Pharmaceutical Development, May 2006.
3. ICH Q8 (R1) — Pharmaceutical Development Revision 1, November 2007.
4. Juran Jm. Juran on quality by design The new steps for planing quality into goods and services newyork free press 1992 p.no-1-2.
5. US Food and Drug Administration. Guidance for Industry. PAT—A Framework for Innovative Pharmaceutical Manufacturing and Quality Assurance. Pharmaceutical cGMPs,Rockville, Pharmaceutical cGMPs. MD, 2004 Sept., 1–21.
6. Pharmaceutical “Quality by Design” (QbD): An Introduction, Process Development and Applications ,august 21, 2012, Process Validation, Validation Articles.
7. Snee Ronald ,Building a framework for quality by design.
8. Moheb M. Nasr “Quality by Design (QbD) – A Modern System Approach to Pharmaceutical Development and Manufacturing –FDA Perspective/ fda.gov
9. Delasko, J.M., Cocchetto, D.M., Burke. L.B., Target Product Profile: Beginning Drug Development with the End in Mind. January/February, Issue 1, 2005, fdli.org
10. Food and Drug Administration CDER. Draft Guidance for Industry and Review Staff: Target Product Profile- A Strategic Development Tool (March 2007).
11. Food and Drug Administration Office of Generic Drugs. Model Quality Overall Summary for IR Product, fda.gov/ cder/ ogd/OGD_Model-QoS_IR_Product.pdf (Accessed March 31, 2006)
12. US Food and Drug Administration. Guidance for industry: Q8 Pharmaceutical Development, US Department of Health and Human Service, FDA, Rockville, MD, May 2006
MORE……….
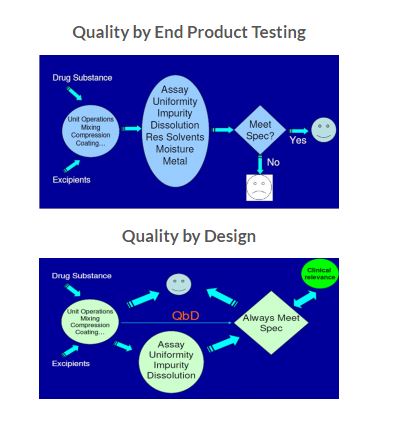
Pharmaceutical industry is constantly searching the ways to ensure and enhance product safety, quality and efficacy. However, drug recalls, manufacturing failure cost, scale up issues and regulatory burden in recent past produce huge challenge for industry. In traditional, the product quality and performance are predominantly ensured by end product testing, with limited understanding of the process and critical process parameters. Regulatory bodies are therefore focusing on implementing quality by design (QbD), a science based approach that improves process understanding by reducing process variation and the enabling process-control strategies.
Instead of relying on finished product testing alone, QbD provides insights upstream throughout the development process. As a result, a quality issue can be efficiently analyzed and its root cause quickly identified. QbD requires identification of all critical formulation attributes and process parameters as well as determining the extent to which any variation can impact the quality of the finished product [1-3].
In the area of pharmaceutical quality; Food and drug administration (FDA) announced proposed amendments to “Current Good Manufacturing Practices” (cGMP) in 2002, with an emphasis on establishing a 21st century outlook on pharmaceutical manufacturing in order to establish a more systematic science and risk based approach to the development of pharmaceutical products. The initiation of the cGMPs for the 21st Century and the publication of the Process Analytical Technology (PAT) guidance in 2004 by the FDA gave the way for the modernization of the pharmaceutical industry. After that, ICH (International Conference on Harmonization) discussions in July 2003 (Brussels) agreed a consensus vision to develop a harmonized pharmaceutical quality system applicable across the life cycle of the product emphasizing an integrated approach to risk management and science. All the major objectives with regard to quality issues are being addressed by the ICH guidelines. The three ICH guidelines which throw light upon quality-by-design and related aspects include Q8 Pharmaceutical development, Q9 Pharmaceutical risk management and Q10 Pharmaceutical Quality systems. In fact, the ICH guideline Q8 is sub-divided into two parts: part one deals with pharmaceutical development and Part II is the annex to the guideline which states the principles for Quality-by-Design. According to ICH Q8(R2) guideline, Quality by Design (QbD) is “A systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and Process control, based on sound science and Quality Risk Management”[4-6].
QbD describes a pharmaceutical development approach referring to formulation design and development and manufacturing processes to maintain the prescribed product quality. Guidelines and mathematical models are used to ensure the establishment and use of the knowledge on the subject in an independent and integrated way [7]. In order to initiate a successful QbD program, the first step is to identify those process parameters that are essential to product quality and develop well – validated analytical methodologies to monitor those parameters. The objective of this review article is therefore to provide a comprehensive understanding on various aspects of QbD, along with addressing the concerns related to its implementation.
Key characteristics of QbD [8-9]
- A tool for focused & efficient drug development
- Dynamic and systematic process
- Relies on the concept that Quality can be built in as a continuum
- It is applicable to Drug Product and Drug Substance development (chemicals / biologics)
- It is applicable to analytical methods
- Can implemented partially or totally
- Can be used at any time in the life cycle of the Drug
- Always encouraged by Regulators.
Benefits of QbD [10-12]
- Eliminate batch failures
- Minimize deviations and costly investigations
- Avoid regulatory compliance problems
- Empowerment of technical staff
- Efficient, agile, flexible system
- Increase manufacturing efficiency, reduce costs and project rejections and waste
- Build scientific knowledge base for all products
- Better interact with industry on science issues
- Ensure consistent information
- Incorporate risk management
- Reduce end-product testing
- Speed-up release decision
Key elements of QbD
ICH Q8: Pharmaceutical Development discusses the various elements of quality by design. These in combination with the enablers form the fundamental basis for the QbD approach to development. Figure 1 provides a pictorial representation of the typical elements of QbD.
It involves the following key elements during pharmaceutical development
- Define the Quality Target Product Profile
- Identify the Quality Attributes
- Perform a Risk (Assessment) Analysis
- Determine the Critical Quality Attributes and Critical Process Parameters
- Determine the Design Space
- Identify a Control Strategy

Fig 1. Overview of QbD.
Quality Target Product Profile (QTTP)
According to ICH Q8(R2), QTTP is “Prospective summary of the quality characteristics of a drug product that ideally will be achieved to ensure the desired quality, taking into account safety and efficacy of the drug product”. Basically it is a tool for setting the strategy for drug development. Recently QTTP is widely used in development planning, clinical and commercial decision making, regulatory agency interactions, and risk management.
It is the quality characteristics that the drug product should possess in order to reproducibly deliver the therapeutic benefit promised in the label. The QTTP guides formulation scientists to establish formulation strategies and keep the formulation effort focused and efficient. QTPP is related to identity, assay, dosage form, purity, stability in the label. For example, a typical QTPP of an immediate release solid oral dosage form would include
– Tablet Characteristics
– Identity
– Assay and Uniformity
– Purity/Impurity
– Stability, and
– Dissolution
It is important to acknowledge that QTPP should only include patient relevant product performance elements. For example, tablet density or hardness may be included as a specification for process monitoring but may not be included in QTPP. Also, if particle size is critical to the dissolution of a solid oral product, then the QTPP should include dissolution but not particle size[13-14].
Critical quality attributes (CQAs)
Once QTPP has been identified, the next step is to identify the relevant CQAs. A CQA is defined as “A physical, chemical, biological or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality”.
CQAs are generally associated with raw materials (drug substance, excipients), intermediates (in-process materials), and drug product. Drug product CQAs are the properties that are important for product performance, that is, the desired quality, safety, and efficacy (Fig.2).
This indicates that CQAs are subsets of QTPP that has a potential to be altered by the change in formulation or process variables [14-15]. For example, QTPP may include additional quality attributes of the drug product such as strength and dosage form, which are not the part of CQA as it will not change during drug development process. However, QTTP attributes such as assay, content uniformity, dissolution, and permeation flux will also be a part of CQA as they may be altered by formulation or process variables. For example, the CQAs of drug substance and drug product are enlisted in Table 1.
Identification of CQAs is done through risk assessment as per the ICH guidance Q9. Prior product knowledge, such as the accumulated laboratory, nonclinical and clinical experience with a specific product-quality attribute, is the key in making these risk assessments. Such knowledge may also include relevant data from similar molecules and data from literature references. Taken together, this information provides a rationale for relating the CQA to product safety and efficacy [16].

Fig. 2. Decision Tree to Decide CQAs.
Table 1. Typical CQAs for drug substance and drug products.
|
For Drug Substance (chemical) |
For Drug product (tablet) |
|
|
Quality risk management (QRM)
The FDA defines a Risk Management as, a strategic safety program designed to decrease product risk by using one or more interventions or tools. It is systematic process for the assessment, control, communication and review of risks to the quality of the drug product across the product lifecycle [17]. Overview of a typical quality risk management process is given in Fig. 3.
The ICH Q9 guideline: Quality Risk Management provides a structure to initiate and follow a risk management process. The relevant tools of QRM are as follows:
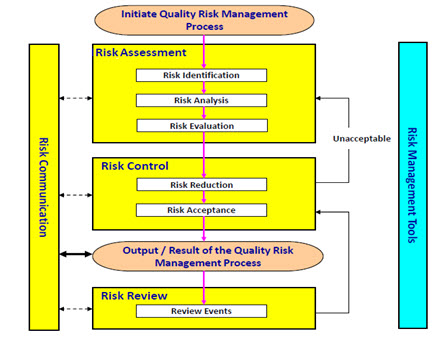
Fig. 3. Overview of a typical quality risk management process (as per ICH Q9: Quality Risk Management).
Failure mode effects analysis (FMEA)
FMEA is one of the most commonly used risk-assessment tools in the pharmaceutical industry. It is a systematic and proactive method to identify and mitigate the possible failure in the process. Failure modes represent any errors or defects in a process, material, design, or equipment. Once failure modes are established, FMEA tool evaluates the effect of these failures and prioritizes them accordingly. This tool is further advanced with studying criticality of the consequences and providing clear indication of situation.
Failure Mode, Effects and Criticality Analysis (FMECA)
It is the extension of earlier said FMEA tool. Extending FEMA to incorporate an investigation of the degree of severity of consequences, their probabilities of occurrence and their detect-ability is Failure mode, effects and criticality analysis. In FMECA, each failure mode of the product is identified and then evaluated for criticality. This criticality is then translated into a risk, and if this level of risk is not acceptable, corrective action must be taken. This can be utilized for failure and risk associated with manufacturing processes. The tool can also be used to establish and optimize maintenance plans for repairable systems and/or contribute to control plans and other quality assurance procedures.
Fault tree analysis (FTA)
This tool assumes failure of the functionality of a product or process. The results are represented pictorially in the form of a tree of fault modes. This can be used to investigate complaints or deviation in order to fully understand their root cause and ensure that intended improvement will resolve the issues and not cause any other different problem.
Hazard analysis and critical control points (HACCP)
HACCP provides detailed documentation to show process or product understanding through identifying parameters to control and monitor. The definition of hazard includes both safety and quality concern in a process or product. It involves hazard analysis, determining critical control point, establishing critical limit, establishing a system to monitor critical control point and establishing a record keeping system. This might be used to identify and manage risk associated with physical, chemical and biological hazards.
The output of a risk assessment may be a combination of quantitative and qualitative estimation of risk. As part of FMEA, a risk score or Risk Priority Number (RPN) may be assigned to the deviation or to the stage of the process that is affected; this helps to categorize the deviation. RPN is calculated by multiplying Probability (P), Detectability (D) and Severity (S),which are individually categorized and scored. Rating scales usually range from 1 to 5.
RPN = probability score × severity score × detectability score
Where, the score was defined prior to the risk analysis stage. A RPN of < 40 was considered a low risk; a RPN of 40–99 was identified as an intermediate risk; and a RPN of ≥ 100 was defined as a high risk [14, 17].
Determination of Critical Process Parameters
A critical process parameter (CPP) is any measurable input (input material attribute or operating
parameter) or output (process state variable or output material attribute) of a process step that must be controlled to achieve the desired product quality and process consistency. A parameter is critical when a realistic change in that parameter can cause the product to fail to meet the QTPP. Thus, whether a parameter is critical or not depends on how large of a change one is willing to consider. Thus the first step in classifying parameters is to define the range of interest which we call the potential operating space (POS). The POS is the region between the maximum and minimum value of interest for each process parameter. Criteria for identifying critical and non-critical parameters are that a parameter is non-critical when there is no trend to failure within the POS and there is no evidence of interactions within the proven acceptable range (PAR), which is the range of experimental observations that lead to acceptable quality [18]. The different CCPs during tablet manufacturing along with CQAs are given in Table 2.
Table 2. Different critical process parameters with potential quality attributes during tableting.
|
Operations during tableting |
Critical Process Parameters |
Potential quality attributes |
|
Wet granulation |
|
|
|
Drying |
|
|
|
Milling |
|
|
|
Mixing |
|
|
|
Compression |
|
|
|
Coating |
|
|
Design space
ICH Q8(R2) defines design space as “ the multidimensional combination and interaction of input variables (e.g., material attributes) and process parameters that have been demonstrated to provide assurance of quality. Working within the design space is not considered as a change. Movement out of the design space is considered to be a change and would normally initiate a regulatory post approval change process. Design space is proposed by the applicant and is subject to regulatory assessment and approval.”
Design space may be constructed for a single unit operation, multiple unit operations, or for the entire process. Though according to FDA guideline, defining design space is optional since the product and process understanding can be established without a formal design space, nevertheless, such approach can assist to better understanding and attain overall control of a system.
The Design Space is linked to criticality through the results of risk assessment, which determines the associated CQAs and CPPs. It describes the multivariate functional relationships between CQAs and the CPPs that impact them, and should include their linkage to or across unit operations. Such relationships are arrived at by iterative application of risk assessment and experimental design, modeling, as well as the use of literature and prior experience.
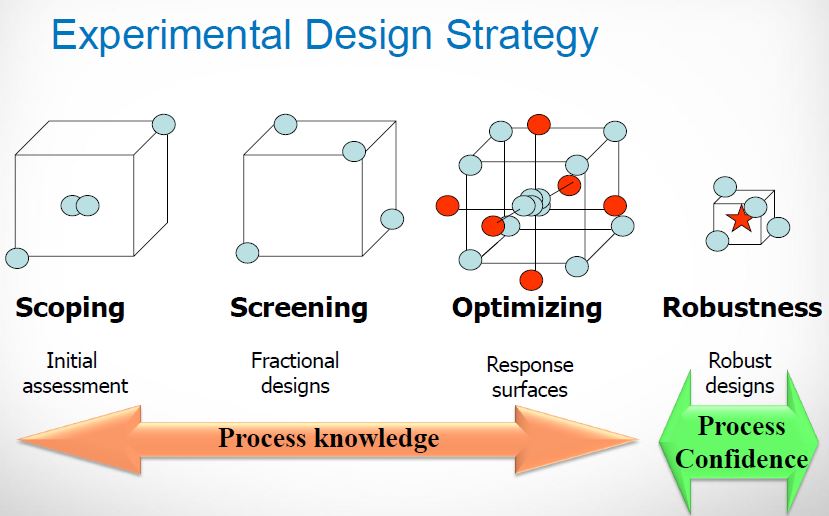
Methods for determining design space included: one-variable-at-a-time experiments, statistically designed experiments, and modeling approaches. Methods for presenting design space included graphs (surface-response curves and contour plots), linear combination of parameter ranges, equations, and models. Alternatively, the design space can be explained mathematically through equations describing relationships between parameters for successful operation [19-21].
Control Strategy
ICH Q10 defines a control strategy as “a planned set of controls derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to drug substance and drug product materials and components, facility and equipment operating conditions, in process controls, finished product specifications and the associated methods and frequency of monitoring and control.” A control strategy normally include input material controls, process controls and monitoring, design space around individual or multiple unit operations, and/or final product specifications used to ensure consistent quality [22, 23]. The finished drug products are tested for quality by assessing if they meet specifications. In addition, manufacturers are usually expected to conduct extensive in process tests, such as blend uniformity or tablet hardness.
A QbD based control strategy for blending process is shown in Fig. 4. Pharmaceutical quality is assured by understanding and controlling formulation and manufacturing variables to assure the quality of the finished product. The end product testing only confirms the quality of the product.
Fig. 4. Example of control strategy for QbD process.
Challenges
Though Quality by design is an essential part of the modern approach to pharmaceutical quality, but Lack of understanding regarding the pharmaceutical process is the cause and also the major limitation for QbD implementation. Pharmaceutical companies are traditionally tuned to care more about the end product, with little emphasis on the science-based understanding of the process involved. The majority of pharmaceutical companies feel that there is a need for a more easy guidance on how to actually implement QbD. Companies wanted clarification from FDA on QbD terminologies, acceptable methods, criteria to select and deselect critical quality attributes, standards by which to judge adequacy of controls, and criteria for analytical method substitution [23, 24]. 10 key challenges are the most problematic for QbD adoption. These challenges are evaluated by their relevancy against different drug types as well as different levels of adoption.
The first four challenges occur within companies:
- Internal misalignment (Disconnect between cross functional areas, e.g., R&D and manufacturing or quality and regulatory)
- Lack of belief in business case i.e. there is a lot of uncertainty over timing of and investment requirements for QbD implementation.
- Lack of technology to execute (e.g., Difficulty managing data, limited understanding of Critical Quality Attribute (CQA) implications)
- Alignment with third parties (i.e., How to implement QbD with increasing reliance on suppliers and contract manufacturers?)
The next six challenges are directly related to the regulatory authority:
- Inconsistency of treatment of QbD across regulatory authority
- Lack of tangible guidance for industry
- Regulators not prepared to handle QbD applications
- The way promised regulatory benefits are currently being shared does not inspire confidence
- Misalignment of international regulatory bodies
- Current interaction with companies is not conducive to QbD
It is accepted that the challenges and concerns associated with the implementation of QbD can only be resolved if there is efficient communication between the industry and the regulatory bodies.
CONCLUSIONS
QbD is increasingly becoming an important and widely used technique in pharmaceutical product development. While QbD is most effective when it is employed at a product/process design level, it should also be accomplished in the manufacturing and quality assurance environments. Implementing QbD concept in product development provide quality medicines to patients, production improvements to Manufacturers with significantly reduced batch failures and drug regulatory bodies will have greater confidence in the robust quality of products. This approach allows the establishment of priorities and flexible boundaries in the process. As such QbD is becoming a promising scientific tool in quality assurance in pharmaceutical industry.
REFERENCES
1. Woodcock J, The concept of pharmaceutical quality. American Pharmaceutical Review 2004;7: 10–15.
2. Lionberger RA, Lee LS, Lee L, Raw A,Yu LX. Quality by design: Concepts for ANDAs. The AAPS Journal 2008; 10: 268–276.
3. Looby M, Ibarra N, Pierce JJ, Buckley K, O’Donovan E, Heenan M. Application of quality by design principles to the development and technology transfer of a major process improvement for the manufacture of a recombinant protein. Biotechnology Progress 2011;27:1718-29.
4. Q9: Quality Risk Management. ICH Harmonized Tripartite Guidelines. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2006.
5. Q10: Pharmaceutical Quality System, ICH Tripartite Guidelines. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2007.
6. Q8 (R1): Pharmaceutical Development, Revision 1, ICH Harmonized Tripartite Guidelines, International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2007.
7. Yu LX, Pharmaceutical quality by design: Product and process development, understanding, and control. Pharmaceutical Research 2008; 25: 781–791.
8. Gupta A, Fuloria NK. Short review on Quality by design: A new Era of Pharmaceutical drug development. Int. J. Drug Dev. & Res. 2012; 4:19-26.
9. Elliott P, Billingham S, Bi J, Zhang H. Quality by design for biopharmaceuticals: a historical review and guide for implementation. Pharmaceutical bioprocessing 2013; 1:105-122.
10. Nadpara NP, Thumar RV, Kalola VN, Patel PB. Quality By Design (QbD) : A Complete Review. Int. J. Pharm. Sci. Rev. Res. 2012; 17: 20-28.
11. Pohl M, Schweitzer M, Hansen G, Hanna BM, Borman P, Smith K, Larew J, Nethercote P. Implications and opportunities of applying the principles of QbD to analytical measurements. Pharm. Technol. Eur. 2010; 22: 29–36.
12. Eziokwu NV. Quality by design (QbD): manufacturing and product quality of Generics drugs perspective. Journal of Global Trends in Pharmaceutical Sciences 2013;4: 1257-1262.
13. Yu LX, Lionberger R, Olson MC, Johnston G, Buehler G, Winkle H. Quality by Design for Generic Drugs. Pharmaceutical Technology 2009;33:122-27.
14. Patil AS, Pethe AM. Quality by Design (QbD) : A new concept for development of quality pharmaceuticals. International Journal of Pharmaceutical Quality Assurance 2013; 4: 13-19.
15. Nagar M, Panwar KS, Chopra VS, Bala I, Triv P. Quality by design: A systematic approach to pharmaceutical development. Der Pharmacia Lettre 2010; 2: 111-130.
16. Kumar VP, Gupta NV. A Review on quality by design approach (QBD) for Pharmaceuticals. Int. J. Drug Dev. & Res. 2015; 7: 52-60.
17. Bhattacharya J. Quality Risk Management –Understanding and Control the Risk in Pharmaceutical Manufacturing Industry. International Journal of Pharmaceutical Science Invention 2015; 4: 29-41.
18. Chowdary KPR, Ravi Shankar K, Kumar PS. Recent research on QbD approach in formulation development – a review. International Journal of Chemical Science and Technology 2014; 4: 282-292.
19. Wu H, White M, Khan MA. Quality-by-Design (QbD): An integrated process analytical technology (PAT) approach for a dynamic pharmaceutical co-precipitation process characterization and process design space development. Int J Pharm. 2011;405:63-78.
20. Altan S, Bergum J, Pfahler L, Senderak E, Sethuraman S, Vukovinsky KE. Statistical Considerations in Design Space Development (Part I of III). Pharmaceutical Technology 2010;34: 66-70.
21. Altan S, Bergum J, Pfahler L, Senderak E, Sethuraman S, Vukovinsky KE. Statistical Considerations in Design Space Development (Part II of III). Pharmaceutical Technology 2010; 34: 52-60.
22. Trivedi B. Quality by desing (QbD) in pharmaceuticals. Int J Pharm Pharm Sci. 2012; 4:17-29.
23. Jain S. Quality by design (QbD): a comprehensive understanding of implementation and challenges in pharmaceuticals development. Int J Pharm Pharm Sci. 2013; 6: 29-35.
24. Drakulich, A. Critical challenges to implementing QbD: A Q&A with FDA. Pharm. Technol. 2009; 33: 90–94.





QbD: Lessons Learned From An FDA Filing
Implementing quality by design offers not only a proactive approach to product development, but also the opportunity to streamline your filing process.
Whether you’re a well-established pharmaceutical company with more than 50 years of experience, or you just opened your doors last week, completing your first quality by design (QbD) filing with the FDA is going to be a challenge. After all, if it were easy, everyone would be doing it. QbD promises a more efficient manufacturing process, with quality and safety you can count on every time. Who wouldn’t want that?
 The problem is it isn’t easy, and with the FDA not mandating the systematic approach of QbD, many haven’t veered from their current filing processes. But with so many benefits of using QbD — getting your drug to market quicker, reduced recalls and rejects, minimized post-approval changes — it may be time to start considering the change. The team at Ash Stevens, a fullservice pharmaceutical CMO, learned that implementing QbD does require a significant investment of time and money. Nevertheless, the end result is a more well-defined and carefully planned manufacturing process.
The problem is it isn’t easy, and with the FDA not mandating the systematic approach of QbD, many haven’t veered from their current filing processes. But with so many benefits of using QbD — getting your drug to market quicker, reduced recalls and rejects, minimized post-approval changes — it may be time to start considering the change. The team at Ash Stevens, a fullservice pharmaceutical CMO, learned that implementing QbD does require a significant investment of time and money. Nevertheless, the end result is a more well-defined and carefully planned manufacturing process.
Making The Switch To QbD
If you’re manufacturing pharmaceuticals you cannot compromise quality. Not only does every drug that leaves a manufacturer’s facility do so with that company’s name on it, it also gets delivered to a patient who depends on its efficacy, purity, and safety. So while not every company is implementing a QbD process that is directly in line with current guidance documents, there is always some level of quality and risk assessment taking place.
Ash Stevens has been using some of the tools and principles of QbD for more than 20 years. However, for their first FDA filing, the Ash Stevens team adjusted their current approach to a more structured one, as outlined by the ICH Harmonised Tripartite Guideline for Pharmaceutical Development Q8, a guidance document for QbD. “We were already using many of the tools involved in a QbD filing, such as design of experiment and identifying critical failure points. The guidance just provides a helpful framework to insert our current process into, and it’s one the FDA would like people to use,” says Charles Stankovic, Ash Stevens principal scientist, regulatory affairs and QA. “The idea is to do more work up front to better understand the process, so you don’t have issues down the road that you won’t understand when you’re at a plant scale. We’ve always thought that understanding QbD is essential to carrying out a successful process in the end, and ultimately, we feel that being able to support these filings is important, because of the value it offers our clients.”
For companies currently using the principles of QbD but not formally filing, the transition may be the same, but in an industry that perceives any variation as a risk, change is often not made until it’s mandated. Because the FDA is not enforcing the use of QbD, the method of filing still varies. “Some companies are doing the QbD work, but not necessarily filing it as QbD, because the FDA does not require it,” says Vince Ammoscato, VP of operations at Ash Stevens.
So why are companies shying away from using QbD to file? Stankovic and Ammoscato say there is a very simple explanation — the focus on the cost and time that QbD requires often overshadows its long-term benefits. “Some clients see QbD as a value-added approach. They want to have all of their ducks in a row when they do their filing,” says Stankovic. “Others are skeptical, because the work is done while the project is still in clinical development, a stage where so many of them fail. It’s a harder sell for clients who are skeptical about doing all of this extra work for a project that could ultimately die.”
But if the concern is a loss of time and money, any company not implementing QbD is risking the loss of these resources just as much, if not more. “When you don’t plan up front correctly, and you’re trying to test in quality at the end, you end up wasting a lot of time, which equates to a lot of money,” says Joe Robinson, director of operations of the Midwest region of Commissioning Agents, a company that provides commissioning, qualification, and validation services for large pharmaceutical companies.
| “When you don’t plan up front correctly, and you’re trying to test in quality at the end, you end up wasting a lot of time, which equates to a lot of money.”
Joe Robinson, director of operations of the Midwest region of Commissioning Agents |
 |
The Cost Of Quality
New equipment for statistical support added a significant cost for Ash Stevens during the company’s QbD project. “For the design of experiment studies, we added laboratory equipment, including an automated reactor to do our parametric study,” explains Stankovic. “This gave us an opportunity to understand our processes better, from both an efficiency and safety perspective.”
In addition to these costs, which included statistical software, Ammoscato says they also consulted a statistician to help with the design and evaluation of the design of experiment (DoE) studies. “If someone doesn’t have experience in this area, they should get additional training or hire a consultant. To start on your own and understand the guidance is very difficult,” he explains. “It’s good to collaborate with people who have prior direct experience with QbD.”
Stankovic says that an indirect cost of QbD is the time spent developing and analyzing processes. “We spend more time collaborating with teams doing risk assessments and other aspects of the guidance. There’s a cost to this, but the benefit is you have multiple experts in the same room looking at the process in great detail,” he explains. More time is also spent developing the documentation for the process, as well as the statistical training for scientists.
QbD Doesn’t Have To Be Complex
In addition to the concerns about how much time and money will need to be invested in order to implement QbD, many companies are also concerned with how complex the QbD process can become. Robinson says that even though QbD filing processes require manufacturers to think about the entire manufacturing process ahead of time (unlike current processes that don’t include testing until the end), they don’t always necessitate the lengthy risk assessment process that so many assume it does. “Obviously, you want to minimize the risk, but sometimes you can’t always mitigate everything, because you may inadvertently add other risks,” says Robinson. “Quality by design is intended to make people think about their projects up front and plan better. It may not require them to do a conventional formal risk assessment. It’s basically sitting down and looking at a project from the science side of things and trying to figure out what the critical quality attributes are and what can be done to control those.”
Critical process parameters, or CPPs, are a major component of the QbD process that drive the complexity of any project. Stankovic provides an overview of the process he and his team learned from the ICH (International Conference on Harmonisation) guidance. “The first step is to develop a targeted product profile, or TPP, which describes the intended use of a drug and how it’s going to be delivered,” says Stankovic. “From there, you define the critical quality attributes (CQAs) of the TPP. Then you identify the critical process parameters, which can affect the CQAs,” explains Stankovic. “The process can be as simple or as complex as you want it to be.”
When it comes to outsourcing, flexibility is an important and necessary quality for any partner to possess, but it is especially important when implementing QbD because of the need for both parties to coordinate their processes. “While we may come into the process with different ideas about what QbD is, we have to ultimately agree on what it’s going to mean to the project,” explains Stankovic. “After that, we’re fully aligned.” Ammoscato adds that it’s a collaborative process, and as long as the client’s requests do not compromise Ash Stevens’ standard operating procedures and policies, they will remain fully aligned from beginning to end.
 Several resources are available that provide QbD filing guidelines. While Ash Stevens referenced the ICH Q8, there are also Q9 and Q10 guidances available. In addition, the FDA published a document in January 2011 titled Guidance for Industry on Process Validation: General Principles and Practices. “QbD is more of a mindset than a specific checklist of activities,” explains Stankovic. “There are some guidelines and details, but how you do it is up to you. For the projects we’ve been involved with, each case is very different, but they all have the same overall goal — improving your understanding of the process by starting with the end in mind.”
Several resources are available that provide QbD filing guidelines. While Ash Stevens referenced the ICH Q8, there are also Q9 and Q10 guidances available. In addition, the FDA published a document in January 2011 titled Guidance for Industry on Process Validation: General Principles and Practices. “QbD is more of a mindset than a specific checklist of activities,” explains Stankovic. “There are some guidelines and details, but how you do it is up to you. For the projects we’ve been involved with, each case is very different, but they all have the same overall goal — improving your understanding of the process by starting with the end in mind.”
The Challenge Presented By Continuous Process Qualification
One of the biggest challenges faced by the team at Ash Stevens was making the adjustment to continuous process qualification and what that would mean once the concept was applied. “Since this aspect of the guidance is not yet tested and not very well understood, there is still much uncertainty as to how ideas such as design space will play out with the FDA regarding post-approval changes,” explains Stankovic.
Robinson explains that this concern is not uncommon and that many people are intimidated by the possibility of having to file changes, because they believe this reflects poorly on them in the eyes of the FDA. In reality, Robinson says filing updates to your process shows the FDA that you care about the quality of your product. “They don’t want you to think that once you’re done, you’re done. Quality by design is about thinking ahead and trying to improve and make your product better,” says Robinson. “If there are risks out there, and you find a better way to mitigate them and control parameters differently, there’s nothing wrong with updating your application and doing that.” He adds that these changes are typically approved. “The FDA wants you to be smarter these days. They like seeing you apply forward thinking because that’s ultimately the goal of QbD,” adds Robinson.
Should The FDA Mandate QbD?
Stankovic and Ammoscato believe the time and money invested in adopting QbD practices has been well worth it. However, for companies that don’t have the necessary resources available, it may be hard to justify investing in QbD until the FDA mandates it. Although rumors have swirled in the past about an approaching regulation, there is no indication this will happen any time soon. Despite this, the benefits of implementing QbD are clear. “By applying QbD principles, a company has a better knowledge of its product and what effect any changes would have. This allows them to better control their process, which leads to a better product in the end. That is the whole reason for it — to deliver a better and safer product to the patient,” explains Robinson. “Following QbD guidelines can actually help streamline the process to get the product to market. This is a benefit for both the company and the patients.”
| “QbD is more of a mindset than a specific checklist of activities.”Charles Stankovic, principal scientist for regulatory affairs and QA, Ash Stevens |  |
For CMOs such as Ash Stevens, the application of QbD is also a draw for clients. “If a client is looking at QbD, and we have experience with the QbD process from start to commercialization, it’s obviously going to be an advantage,” says Ammoscato. “It’s always good to be at the leading edge of the newest guidance.”
 Stankovic adds that even though the FDA isn’t requiring QbD, they’re certainly looking for it and happy when they see it in use. “When we had our pre-approval inspection, it was clear the inspectors were glad to see we were using a QbD approach, we were using all of the right terminology, and we had all of the correct reports and documentation,” he explains. “They started with our project validation master plan and used that as a guide to find all of the associated documentation, which made their review faster. Overall, they were satisfied with the approach and the way it was applied.”
Stankovic adds that even though the FDA isn’t requiring QbD, they’re certainly looking for it and happy when they see it in use. “When we had our pre-approval inspection, it was clear the inspectors were glad to see we were using a QbD approach, we were using all of the right terminology, and we had all of the correct reports and documentation,” he explains. “They started with our project validation master plan and used that as a guide to find all of the associated documentation, which made their review faster. Overall, they were satisfied with the approach and the way it was applied.”
Until the FDA pushes through a mandate requiring the use of QbD, many companies will continue to stick to the traditional approach or even apply a hybrid. But as more companies adopt Qbd, it will become the unspecified standard, as well as the expectation. Despite the issues with cost and time, good business can only be attained through great science, and QbD could help a company find the perfect balance.
//////////
101,659 total views, 1 views today
