
While all of the original points remain challenges during API drug development, would include the rise of data analytics and the adoption of Quality by Design (QbD). Others, such as weak impurity profiles, proper route selection and weak impurity characterization remain front-and-center challenges for many pharma companies today.
- QbD: All Aboard.
QbD isn’t going away – quite the contrary, it’s just starting to make its presence felt. Since Merck’sJanuvia became the first product approved based on a QbD application in 2006, Quality by Design (QbD) has been setting itself deep into the pharma development and manufacturing industry – and to largely positive effect. (Our experience at Neuland with QbD has been positive, as well).At its core, QbD aims to maximize product safety & efficacy while improving the underlying manufacturing process. Quality by Design is proving itself to be an effective framework, and pharma sponsors are increasingly looking for contract research & manufacturing firms who are QbD-capable.
- Data – it ALL Matters.
We are deep into the era of Big Data analytics. The amount of data we have available to us is enormous, and growing – sometimes uncontrollably.But data is your friend, and it holds the answers to critical regulatory decisions that companies face. Data analytics can help deliver accurate, rapid decisions during discovery, development, manufacturing, clinical stages and ultimately commercial post-market.Given that data management & analytics are increasingly essential to the pharma industry, they must be given strong consideration across the entire spectrum of the drug lifecycle.
These last four challenges from my previous list still apply:
- Route Selection
Choosing the right synthesis route still remains a top priority, given the cost of “getting it wrong” (or – and perhaps more likely – developing a longer, less cost-effective route with potential challenges).Chemical synthesis route scouting is intended to stave off production problems – genotoxic or other impurities, overuse of chemicals and reagents (and the accompanying EHS issues that may arise), manufacturing times & capacities…route scouting impacts a number of critical points in the manufacturing chain, and can have an outsized impact on them.
- Incomplete Stability Data
New APIs mean a lack of stability data, and that doesn’t bode well for regulatory success…or established expiration dates. Make sure you’ve considered this, and your supplier/partner can handle any studies that need to occur.If outsourcing stability testing to a third party, it’s important to ensure the smooth transfer of data and knowledge from your supplier to the testing provider. Make sure stability studies are performed in accordance with ICH guidelines and other protocols.
- Weak Impurity Characterization
Analytical and other testing methods must be fully developed and validated. Remember: identifying, characterizing and synthesizing your API is crucial…but so is identifying, characterizing and synthesizing the impurities that typically surround it. Bottom line: the impurity profile matters to your API’s regulatory success.
- Problems with Genotoxic Impurities
Genotoxic impurities continue to be a major source of regulatory headaches. They are often cited as one of the reasons clients approach Neuland for alternate route scouting & selection services. But GIs are more than just a manufacturing challenge: analytical methods need to be highly-sensitive to detect them. And while many genotoxic impurities can be avoided, often your manufacturing partner will need to focus on controlling them – whether by adjusting environmental controls, altering purge strategies, using preservatives or another means.
These are some of the top API regulatory challenges we see in the industry today – from the rise of QbD &data analytics to continued challenges characterizing the stability & impurities of new chemical entities.

Impurity isolation and sample purification
During a manufacturing process or a stability study for an Active Pharmaceutical Ingredient (API) or drug product, a sample may be found to contain impurities. These impurities might occur as degradation products, as intermediates/process impurities originating from the synthetic route, as an unwanted by-product of side-reactions or due to contamination of the original sample. Such impurities can arise at very low concentrations relative to the API, perhaps as little as 0.05% w/w, but it is important that when discovered they are isolated and identified. The ICH Q3 guidelines for Impurities in New Drug Substances (Q3A(R2)) and for Impurities in New Drug Products (Q3B(R2)) clearly state that degradation products observed during manufacturing and stability studies, conducted at the recommended storage conditions, should be identified when present at a level greater than the identification thresholds given in Tables 1 and 2.
Table 1: Thresholds for Degradation Products in New Drug Substances (Q3A(R2))
| Maximum Daily Dose1 |
Reporting Threshold 2,3 |
Identification Threshold3 |
Qualification Threshold3 |
| ≤2 g |
0.05% |
0.10% or 1.0 mg / day intake* |
0.15% or 1.0 mg / day intake* |
| >2 g |
0.03% |
0.05% |
0.05% |
1. The amount of drug substance administered per day
2. Higher reporting thresholds should be scientifically justified
3. Lower thresholds can be appropriate if the impurity is unusually toxic
*whichever is lower
Table 2: Thresholds for Degradation Products in New Drug Products Reporting Thresholds (Q3B(R2)
| Maximum Daily Dose1 |
Reporting Threshold 2,3 |
Identification Thresholds2,3 |
Qualification Thresholds2,3 |
| ≤1 g |
0.1% |
– |
– |
| >1 g |
0.05% |
– |
– |
| <1 mg |
– |
1.0%* or 5 µg TDI* |
– |
| 1 mg -10 mg |
– |
0.5%* or 20 µg TDI* |
– |
| >10 mg-2 g |
– |
0.2%* or 2 mg TDI* |
– |
| >2 g |
– |
0.10% |
– |
| <10 mg |
– |
– |
1.0%* or 50 µg TDI* |
| 10 mg – 100 mg |
– |
– |
0.5%* or 200 µg TDI* |
| >100 mg – 2 g |
– |
– |
0.2%* or 3 mg TDI* |
| > 2 g |
– |
– |
0.15% |
1. The amount of drug substance administered per day
2. Threshold for degradation products are either expressed as percentage of the drug substance or as total daily intake (TDI) of the degradation product. Lower thresholds can be appropriate if the degradation product is unusually toxic.
3. Higher threshold should be scientifically justified
*whichever is lower
When the impurities are known impurities of the API, they can often be identified via retention time match and spectral or mass spectrometry (MS) confirmation. However, in most cases, when unknown degradation products are found, they require isolation and identification. On occasions, quantities as low as 5 mg can be sufficient for structural elucidation but it is preferable to isolate around 20 – 40 mg of impurity for full structural elucidation.
Preparative LC
The technique best suited to isolating impurities is preparative liquid chromatography (LC), using low or high-pressure columns. The technique requires loading of a preparative-scale LC column with repeated doses of the sample, and collecting fractions either using known time intervals or mass-based fraction collection (using the mass of the molecule you are collecting). This allows isolation and concentration of sufficient impurity for identification purposes using highly sophisticated techniques, such as nuclear magnetic resonance (NMR) spectrometry, Fourier transform infrared spectroscopy (FT-IR) and MS, to help identify the chemical impurity. Of course, there are some challenges with this approach. The process of isolating and concentrating the impurity is not always as straightforward as it may sound above.
The specific choice of preparative LC method will rely on what is already known about the chemical structure of the drug molecule: its known or anticipated impurities, stability and solubility data, earlier used chromatographic methods and much, much more. This pre-knowledge will help determine what sort of chromatography might be best suited for the isolation.
It is important to optimise the chromatography at an analytical scale before scale-up and increasing the flow rate for preparative LC. This can avoid wasting large quantities of expensive solvents, where required. When scaling up the chromatography from analytical to preparative LC, an increase in both the sample concentration and the flow-rate of the mobile phase is required. A sensible approach is to increase the sample concentration before increasing the mobile phase flow rate, and to optimise the selectivity and resolution between the peak of interest and other peaks at analytical scale. However, keep in mind that the sample loading capacity of any column must not be exceeded when performing these optimisation experiments. This allows you to scale up a method that is already optimised before using large quantities of solvent on a Prep LC.
Clearly, the aim of this work is to enable sufficient resolution to minimise issues from co-eluting compounds and obtain as pure an impurity as possible. Hence, wherever possible, develop an LC method that does not use additives in the mobile phase. If this is not possible, the additive used must be easily removable. The mobile phase must also be able to solubilise the sample at the higher concentrations used for preparative LC, and be suitable for the NMR, MS or FT-IR analysis that will be used after isolation. Finally, the mobile phase will ideally be of low viscosity, to stop high back pressure building up in the system.
After performing the LC separation and fraction collection, the enriched or purified impurity has to be recovered from the mobile phase without degradation. This is not always straightforward as the impurity may be unstable under the conditions of isolation.
Performing the isolation
It is good practise to investigate both the pooled fraction and the recovered material, so that should there have been any degradation of the unknown impurity during the recovery process, it can be detected. Thereafter, the recovered material can be subjected to the next LC separation or, following the final purification step, submitted to analytical investigation. Once again, the quality of solvents used for these separations is critical, especially if evaporation is to be applied for recovery. Impurities from the solvent could contaminate the isolated impurity, and present difficulties in the elucidation of its structure. The use of highly pure HPLC-grade solvents at the stage of final purification can help to alleviate the risk of such contamination. If solvent recycling is used, care must be taken to ensure collected fractions are not contaminated with co-eluting compounds. It is also important to ensure that the mobile phase solvent is volatile if evaporation is being used as the final isolation step. Water can often be successfully removed by salting out and evaporating the organic layer only, assuming the compound of interest is preferentially soluble in the organic media. This can dramatically reduce the evaporation time.
After the impurity isolation is complete, it is possible to collect milligram quantities of material, which should be enough for the identification techniques mentioned above. It is also important to confirm the purity of the isolated material using high-resolution analytical columns (and more than one separation technique may be required) to confirm the purity of the isolated material.
Alternative use
Preparative HPLC is also suitable for isolating reference standard-quality material. Again, this is performed by loading the sample onto the column and collecting fractions within a narrow band of the eluted sample peak, where the target molecule should be at its purest. Depending on the sample and size of column used, a few grams of reference standard can be produced in this manner. The identification techniques of NMR, FT-IR and MS can be used to aid in the characterisation of this material (along with other techniques such as Karl Fischer, HPLC, heavy metals, sulphated ash) and subsequently established as a Primary Reference Standard. Where biopharmaceutical molecules are purified, alternative techniques such as amino acid analysis, peptide mapping (using LC-MS), gel electrophoresis etc. may be used to confirm purity and identity.
The approach is appropriate for small and large molecules, depending on the nature of the columns used, meaning it has applications for both traditional pharmaceutical and biopharmaceutical customers.
Conclusion
Impurity isolation, identification and sample purification are essential aspects of pharmaceutical development, and these services are an important part of the support that RSSL can offer pharmaceutical customers, for both existing compounds and new Chemical/Biological Entities.

Method development – why it matters to get it right
Method development – why it matters to get it right
In an industry that is seeing increasing levels of outsourcing, the contract research organisation (CRO) needs to have proven experience in both the pragmatism and flexibility of method development and a regulatory background in validation.
Pharmaceutical companies are focusing on achieving ever shorter times of drug to market, so it is vital that a tailored, pragmatic approach is adopted when conducting method development for active pharmaceutical ingredients (API) or drug products (DP).
Although methods require a high degree of robustness, the overall strategy should encompass full evaluation of the regulatory requirements applicable to the particular phase of the drug lifecycle; this is pivotal to ensure a successful regulatory submission, whereby the applicant must demonstrate suitable validation of all methods used to support the filing.
Successfully developed (and validated) analytical methods can reduce overall turnaround times from preclinical to commercial release. Methods should have the desired flexibility built in during early stages to allow easy translation from API to DP, thus potentially reducing costs throughout the product lifecycle.
Method development strategy
Reliable and reproducible analytical methods are essential throughout the pharmaceutical development process and need to be capable of measuring potency, purity and stability of the final drug product.
Although method development activities are applicable to a variety of analytical techniques, this article focuses on HPLC.
The development strategy is summarised in Figure 1 and is often cyclical in nature depending on the findings throughout the development.
A number of questions should be posed at the outset (see Figure 2), including (a) the intended method outcome, (b) the type of matrix the product is to be extracted from and (c), the intended presentation and dose.
Consider the scenario where a reverse phase (RP) HPLC method is required for assay and related substance determination of an API which will subsequently be formulated into a hard gelatin capsule.
Selecting appropriate samples for method development is paramount; they should provide a ‘worst-case’ scenario in terms of reflecting all potential impurities. This will ensure that the method is specific and stability-indicating, i.e. that all known related substances are well resolved from one another and the active peak. Samples should ideally be impure development batches, representative of the final synthetic route (API) and/or manufacturing process (DP). The use of mother liquors, stressed samples, filtrates and stability samples is also recommended.
Assuming a solubility screen has been performed to determine a suitable injection solvent, the first step involves evaluation of analyte chemistry. This includes scrutiny of any potential ionisable groups and basic functionality, together with an evaluation of the pKa data, to determine if pH control is necessary.
Appropriate column technologies should then be selected for initial screening. Consideration should be given to the potential for secondary retention arising from interaction between acidic silanols within the column stationary phase and basic moieties of the API molecule. This could manifest itself as broad, tailing peaks. Adaption of a combination of column chemistry, pH control and addition of a low level modifier may assist in reducing this secondary retention.
Initiation of the development would typically include the use of scouting gradients using a simple mobile phase composition (e.g. acetonitrile/water). A ‘keep it simple’ approach is always advisable to maintain future robustness of the method. Scouting gradients offer a number of advantages in the early stages of the development enabling potentially wide-ranging polarities to be suitably resolved as well as eluting the more non-polar components in a reduced run-time.
It is pivotal from the early stages that method flexibility/robustness is maintained in order to encompass any changes that may occur with the dose and/or the type of presentation. For optimisation of an API assay/related substances method, whilst it is ideal to have as short a run time as possible, removing too much redundant baseline leaves far less scope for future synergy; if/when the API is formulated into DP, the presence of multiple excipients could pose issues if the API method is refined too much.
For compounds with a suitable chromophore, evaluation of the UV spectral profiles for actives and key related substances should be performed. There are fundamental criteria that should be considered as this could impact upon overall robustness of the method.
Figure 3 illustrates the UV spectra for an API and its main impurity. When selecting a detection wavelength, the primary focus would be around maximising sensitivity. At first glance this may suggest that to achieve maximal sensitivity, a detection wavelength of 260nm should be selected since this coincides with the λmax of the API. Alternatively, 220nm could be selected (although this would only give approximately half of the sensitivity for the API).
Areas of the UV curve to avoid are those which sit on a sharp incline or decline since at these regions, only very small changes in UV output could lead to significant changes in peak response, potentially leading to a non-robust method. Therefore, in order to collect both the API and impurity peaks, much closer inspection of the UV curves would be needed; the wavelength selected should not only aim to give maximal response and sit on a shallow point of the slope, but also represent a point whereby responses of active and impurity are closely matched, essential to allow related substances to be collected as area%. Scrutiny of the above suggests a wavelength of 240nm would satisfy these criteria. Further refinement in sensitivity could then be sought via manipulation of solution concentration and/or injection volume.
If synergy in the API and impurity response is not achievable, an alternative joint wavelength could be used, however, relative responses between active/impurity should be calculated. If there is no possibility of a compromise with a single joint wavelength, multiple wavelengths could be used.
Sample preparation is crucial in building a platform for the overall method development process. There are a number of considerations that need to be assessed. In comparison to establishing the chromatographic conditions (Figure 1), insufficient emphasis is often placed on optimising the sample preparation. In DP method development this is often underestimated and can ultimately lead to a less than robust analytical procedure longer term.
The sample preparation should be as simple as possible. A method should not only be fit for successful validation and transfer, but also able to robustly measure key stability characteristics to support shelf-life evaluation.
When preparing a sample solution, a decision needs to be made with regards to the number of dosage units incorporated: this is driven by the need to obtain a suitable sample solution concentration (within solubility limits of the active/impurities), optimisation of column loading (in conjunction with injection volume) to obtain a peak that is within linear range of the detector and provide adequate sensitivity of related substances.
All factors have to be balanced with the need to take a representative number of units, essential to achieving a robust method as it will reduce the impact of any fill weight bias that may skew assay results. Additionally, taking a hard gelatin capsule as an example, the sampling method needs to be carefully considered. For example, transfer the entire capsule (shell and contents), or simply empty the contents (with washings), or perhaps take a representative weighing of the bulk fill? It is preferential to adopt as simple a sample preparation as possible, so the first option would be preferable.
Where possible, lengthy dilution steps should be avoided to minimise errors, maximise recovery and save analytical time. Adjustment of injection volume and UV wavelength could be used as alternative options when refining the column loading.
Another potential area for caution when dealing with high levels of excipients in volumetric analysis is the impact of excluded volume: this can occur if the mass of powder blend taken into a volumetric flask is significant enough to displace volume that would otherwise be occupied by sample solvent. In such instances, consider the addition of a fixed volume of diluent as opposed to diluting up to volume in a flask. Any issue with excluded volume would tend to manifest itself as higher than expected assays due to the lower sample solvent volume.
Caution should also be exercised when bulking the contents of capsules and then taking a weighing as, for early-phase products where the formulation remains in the ‘optimisation’ phase, segregation of the components may occur leading to errors with assay results.
Throughout development, all findings should be continually evaluated to identify parameters that are particularly susceptible to minor adjustment, ensuring that these are experimentally assessed prior to the validation phase. Typically, linearity, extraction efficiency and method repeatability should be well understood ahead of planning the validation to reduce any risk to the future robustness of the method (and significant unwanted time and cost).
Scrutiny of the above should also enable a validation protocol to be produced that is far more representative of the specific API/DP.
Conclusion
Having significant previous experience in the area of method development is central in selecting an appropriate CRO; they need to possess the ability to work in a pragmatic, GMP-compliant manner to achieve a solid method that will ultimately support a successful DP filing and also serve to be reliable and robust in its future use.
API Manufacture: Identifying & Controlling Genotoxic Impurities
Since API manufacturing plays a critical role during clinical development and after approval of a drug, familiarity with genotoxic impurities is an important issue for pharmaceutical firms considering outsourced API manufacturing. Understanding how an API producer – or your own firm’s manufacturing and QA people – identify and resolve genotoxic impurities is critical knowledge for virtually all pharmaceutical execs.
Genotoxic Impurities in Small Molecule Development Programs – A Real Risk
Awareness of the potential for genotoxic impurities (GTIs) during the manufacture of APIs has led to strategies for managing the risks of contamination. We recently took part in hosting a webinar (thanks to everyone who attended) on API Development: Risk Evaluation and Control of Genotoxic Impurities in which we explored the risks of genotoxic impurities in small-molecule API development, today’s regulatory landscape and various approaches to managing GTIs.
If you are not directly involved in the identification and minimization of GTIs, it’s still an important issue. The presence of genotoxic impurities is indicative of contamination and lack of control over the manufacturing process – and, yes, sometimes GTIs are unavoidable. More importantly, they present a potentially serious risk to human health.
The Types and Risks of Genotoxic Impurities
There are three types of genotoxic impurities: mutagens, clastogens and carcinogens. These impurities can also be more than one type; for example, a mutagen can change DNA which leads to cancer – making it a carcinogen as well.
Here’s a short description of types of genotoxic impurities and how they can negatively impact human health:
- Mutagen
Mutagens are agents that change a person’s genetic material, typically the DNA. THE RISK: this leads to above-normal levels of mutations, which can cause cancer. Mutagens can cause gene defects as well as changes to the structure & function of proteins in the body, leading to a wide range of diseases and conditions.
- Clastogen
Clastogens can cause sections of chromosomes to be added, deleted or rearranged by breaking the chromosome. THE RISK: cells that aren’t killed during this process can become cancerous. They have also been reported to have an effect on fetal development. As with mutagens, clastogens can be lethal or cause serious disease.
- Carcinogen
Carcinogens are cancer-causing agents that damage the genome or disrupt cellular processes.
Preventing Genotoxic Impurities
The identification of genotoxic impurities requires a strong, focused analytical chemistry program. Analytical development scientists, in particular, should possess an in-depth understanding of degradation chemistry. Analytical chemists should work closely with the teams developing or manufacturing the API to identify, eliminate or minimize impurities.
Here are two common questions we encounter regarding the risks of GTIs during API manufacture:
- Can genotoxic impurities be avoided?
The fact of the matter is GTIs often are avoidable. It might something as simple as a reagent change, altering the synthetic route, or a purification route change to minimize the formation of the impurity. But not all genotoxic impurities can be completely eliminated.
- Impurities are sometimes unavoidable. What can we do to minimize the risk?
Inevitably, there will be genotoxic impurities that can only be controlled – not eliminated – and the focus shifts towards maximizing the removal of the relevant impurity to comply with the practical impurity limits currently being established by various regulatory bodies. There are steps firms can take to control genotoxic impurities during manufacture, including: adjusting environmental controls, altering purge strategies, using preservatives and others.
During the manufacture of an API – especially at clinical development levels – genotoxic impurities present a very real risk of product contamination. Does your API manufacturing partner have strong analytical development scientists focused on the hazards posed by genotoxic impurities?
Genotoxic Impurities – Increasing Vigilance, But Still Some Uncertainty
 A genotoxic impurity (GTI) is a chemical substance that can directly or indirectly damage DNA or chromosomes and induce genetic mutations. Fifteen years ago, there were no specific guidelines for them.
A genotoxic impurity (GTI) is a chemical substance that can directly or indirectly damage DNA or chromosomes and induce genetic mutations. Fifteen years ago, there were no specific guidelines for them.
In 2007, however, general awareness of the risk and consequences of GTIs surged when Roche’s drug Viracept® was accidentally contaminated in a case that quickly became high-profile. Residual ethanol left in a storage tank reacted with acid over a long period of time, creating high levels of ethyl methane sulfonate (EMS) that remained in the product. The EMS levels in the tablets went undetected until patients who took them showed adverse effects.
Since then, vigilance concerning genotoxic impurities has grown, and regulatory standards have emerged. The regulatory standards governing GTIs, however, don’t take long-term therapeutic usage (potentially higher, longer-term thresholds) into account, and some uncertainty exists regarding several aspects of the standards.
Neuland has been at the forefront of research and method development into sensitive, efficient ways to detect low levels of impurities for some time.
As some genotoxic impurities cannot be completely eliminated, emphasis is placed on sufficiently controlling them to comply with the impurity limits set by regulatory bodies.
Threshold of Toxicological Concern
For unusually-potent impurities or those that produce toxic or unexpected pharmacological effects, the detection and quantification limit of the analysis should match the level at which the impurities should be controlled. The Threshold of Toxicological Concern (TTC) is the level at which someone can be exposed to agenotoxic impurity in most pharmaceuticals with minimal risk, balanced with the therapeutic benefits of taking the drug. The TTC for intake of a genotoxic impurity is 1.5 micrograms per day. Low and high limits are case-specific and based on each compound’s toxic potential.
To assess the potential for GTIs to affect drug quality, a proactive, multidisciplinary approach should be used. A highly conservative limit, the TTC level can only be applied singly to individual GTIs when the impurities are not structurally similar.
With multiple structurally-similar impurities that are expected to act by similar genotoxic or carcinogenicmechanisms, total daily exposure should be evaluated in relation to the TTC. Appropriate individual limits should be applied to the sum of all structurally similar GTIs.
How to Control GTIs
To best detect and control GTIs, we recommend using a planned set of controls – derived from current product and process understanding – that ensures process performance and product quality. Of paramount importance: selecting a route that doesn’t use genotoxic-alerting intermediates or reagents – or a combination with the potential to generate GTIs. Controls include:
- parameters and attributes related to the drug substance and drug product materials and components
- facility and equipment operating conditions
- in-process controls
- finished product specifications, and the associated methods
- frequent monitoring and control
Are Current Genotoxic Impurity (GTI) Regulations Too Weak?
Genotoxic impurities were around long before humans first began boiling bark. Our attempts to quantify, understand and control GTI formation, however, is a much more recent phenomena. It’s also a work in progress, and our knowledge of their potential impact continues to multiply.
When it comes to GTIs, the various teams working here at Neuland keep their ears to the ground. It’s an area in which we’re fairly experienced, and it’s also an area – as a complex chemistry synthesis provider – that stimulates both considerable discussion and some concern.
GTIs are broadly regulated by a variety of regulations and industry guidance including:
In the industry, a great deal of confusion has surfaced regarding contradictory standards and lack of clear guidance on emerging issues. Granted, genotoxic impurities have been the subject of furious research over the last ten years, so rapid change in guidance is to be expected as our understanding of GTI formation and clearance continues to expand and regulatory frameworks adjust and adapt to the growing body of knowledge.
Here are some of the thoughts and concerns that I’ve heard regarding the existing standards.
- Consensus seems to be that the Threshold of Toxicological Concern (TTC) concept is strong, but concerns have also been expressed that the maximum daily exposure of 1.5 µg may be overly conservative.
- Current guidelines don’t provide any clear guidance on GTIs found in clinical/investigational medicines, an area where some guidance would be helpful.
- One particular circumstance that requires clarity: if more than a single potential genotoxic impurity is equally likely to be present, which should be used as the control?
- Currently, there is no specific guidance on natural product-derived and herbal medicines.
- Additional guidance on TTC limits for oncology products should be considered since typical lifetime exposure limits can be exceeded.
- It was noted that manufacturing process changes made to existing and novel excipients could result in potentially higher risks.
- No clear GTI analytical methodologies have been established for compound-specific genotoxicity and carcinogenicity.
- There remain “causes for concern” for existing medicinal products or existing monographs.
- The emergence of in-silico techniques for identifying structural alerts (versus the tradtional Ames test) is a testimony to the power of computing, but risk factors can arise with dataset reliability and accuracy.
API Production: Building an Impurity Profile
Impurities and the ICH Limits for Impurities
Impurities are unwanted chemicals produced during normal manufacturing of Active Pharmaceutical Ingredients, or APIs. As you well know, impurities have no therapeutic value and therefore must be controlled. An impurity profile describes the identified and unidentified impurities a new drug contains.
Arising from many sources, the types of impurities include:
Organic
- starting materials or intermediate impurities
- by-products
- degradation products
- synthesis-related
Inorganic
- reagents, ligands, catalysts
- heavy metals (which are further categorized into 3 classes, per ICH Q3D – Guideline for Elemental Impurities)
- other impurities, such as filter aids, charcoal, etc.
Enantiomeric
- This is a compound with the same molecular formula as the drug, which has a different spatial arrangement of atoms within the molecule and is a mirror image that cannot be super-imposed.
Residual solvents, which are divided into the following 3 classes:
| Class I |
Solvents to avoid |
Known human carcinogens, strongly suspected human carcinogens, and environmental hazards. |
| Class II |
Solvents to limit |
Non-genotoxic animal carcinogens or possible causative agents of other irreversible toxicity. Solvents suspected of other significant but reversible toxicities. |
| Class III |
Solvents with low toxic potential |
Low toxic potential for humans; no health-based exposure limit is needed. |
In-process production impurities
- crystallization-related
- stereochemistry-related
- residual solvents: Class I, II and III
- Synthetic intermediates and by-products
- Impurities caused during storage
As you can see, impurities can arise virtually anywhere across the API development and manufacturing process.
ICH limits for impurities have long been established to minimize the risk posed by such contaminants. The ICH limits are:
| Maximum Daily Dose |
Reporting Threshold |
Identification Threshold |
Qualification Threshold |
| ≤2 g/day |
0.05% |
0.10% or 1.0 mg/day
(whichever is lower) |
0.15% or 1.0 mg/day (whichever is lower) |
| ˃2 g/day |
0.03% |
0.05% |
0.05% |
Methods for Isolating Impurities during API Production
A wide range of methods effectively isolate impurities, which allows us to understand where and how the impurities enter the API manufacturing process. With an understanding of the contaminant’s formation in hand, new methods or process modifications can be crafted to minimize and control them.
Here are some of the common methods used in API manufacturing to help control impurities:
- Solid phase extraction, where compounds that are dissolved or suspended in a liquid mixture are separated from other compounds in the mixture.
- Liquid-liquid extraction, in which a compound is transferred from solvent A to solvent B. Both solvents are liquids that form layers when added together, like oil and water.
- Accelerated solvent extraction, a patented technique that uses common solvents at elevated temperatures and pressures to extract solid and semisolid sample matrices, is also sometimes employed. This method reduces extraction time from hours to minutes.
- Supercritical fluid extraction – chemical compounds are extracted using supercritical carbon dioxide instead of an organic solvent.
- Capillary electrophoresis – in which an applied voltage separates ions according to their electrophoretic mobility – determined by the molecule’s charge and viscosity, and the atom’s radius.
- Numerous types of chromatographic methods have also been shown to be effective. In column chromatography, the mixture to be analyzed is placed inside a column containing stationary phase (usually silica gel/alumina or its derivatized form). The liquid is passed through the column by either gravity (known as gravity column chromatography) or by positive air pressure (known as flash chromatography), with molecules moving at different rates. The eluent (solvent) is collected in fractions and analyzed to gauge the success of the separation. The method used to analyze the fractions isgenerally thin-layer chromatography, where a sheet of glass, metal, or plastic is coated with a thin layer of adsorbent material, such as silica or alumina.
- Gas chromatography – volatile components of a mixture are separated by drawing a small amount of the sample into a syringe. After being injected into a gas chromatograph, the gaseous components are pushed onto the column and separated.
- High/ultra performance liquid chromatography – a reservoir holds the solvent. And in high-performance thin-layer chromatography, the capillary action of a solvent and stationary phase is used to separate compounds in a mixture.
we use a number of methods – including combinations of methods – to isolate contaminants. Which method(s) to use generally comes down to the characteristics of both the API and the contaminants present.
Characterization Methods
To control contaminants, it is critical to understand them. Isolated impurities are subject to a number of potential techniques designed to identify what they are – the first step towards developing methods to control their formation.
Several methods can be used to characterize compounds. In mass spectrometry, a mass spectrometer converts molecules to ions so they can be moved by external electric and magnetic fields. In nuclear magnetic resonance (NMR) spectroscopy, electron distribution leads to a chemical shift in the resonance frequency, making it possible to elucidate structure. Raman spectroscopy measures the wavelength and intensity of inelastic light scattered from molecules.
In liquid chromatography-mass spectrometry (LC-MS), an HPLC system separates chemicals on a column, and a mass detector scans the molecules and separates ions by mass. And in LC-MS-NMR, methods are linked, and peaks of interest are identified by NMR and MS.
Validating Impurity Methods
An impurity profile must be validated in order to ensure the integrity, accuracy and repeatability of the methods chosen. In order to validate impurity methods, the following protocol is used:
- specificity
- linearity/range
- accuracy
- repeatability/intermediate precision
- detection limit/quantitation limit
- robustness
While building an impurity profile is a complex process, knowing how to properly build such a profile is essential as drug safety continues to come under increasing scrutiny from both the media and consumers.
How much emphasis does your organization place on impurity profile design?
Quality Control & Assurance Are Constantly Evolving
Sometimes I feel like I come across as a bit too “QA crazy,” but quality in the pharma sense isn’t really something of which you can have too much. From start (order processing and materials procurement) to finish (consignment shipping to customer), every step must be subjected to quality oversight and monitored.
It goes without saying that pharma isn’t an industry that lends itself to flexibility in quality. You don’t hear: “Sorry, toxicity levels were off the charts. We’ll make it up in the next shipment…it’ll all average out.”
An Eye Towards the Future
Quality, as we in the pharma industry know it, is a collection of standards, methods, data and processes. In a post earlier this month, we discussed building API impurity profiles and mentioned some of those standards, methods, etc. But ‘Quality’ goes far beyond that. It is better viewed as an evolving, vibrant process that is subject to constant review as our collective knowledge – and our ability to produce products to standards that improve both health and outcomes – increases.
The evolution of this thinking has led to practices such as Quality by Design and other continuous process improvement measures. Quality has transformed from static, required measures & procedures into an ongoing, improvement-focused philosophy.
Staying Ahead: Delivering Tomorrow’s Quality Today
There are a number of ways in which companies – especially contract manufacturers, who are often subject to numerous regulatory regimes – address quality issues. At Neuland, for example, maintaining a clear understanding of the possible direction of regulations and standards is essential. We believe it is important to implement policy ahead of legislation.
We continuously monitor compliance with cGMP guidelines established by ICH (Q7A), as well as relevant requirements of the U.S. FDA, ICH, EMEA and EDQM. Stringent controls are built-in to our quality programs to ensure each product meets not only all pharmacopeia specifications, but also those of the customer (which are sometimes even more stringent).
Some of the specific techniques Neuland uses to ensure the quality of every product include:
- Sampling and analyzing products and impurities
- In-process tests for production
- Microbiology laboratory in line with international standards
- Sampling, testing and selection of starting materials
- Sampling, testing and selection of all subsequent inputs, intermediates, and finished products
- Analytical methods development & validation
- In-process control of production operations
- Specific internal instrument qualification and calibration programs
- Stability testing of products
- Microbial limit testing & BET testing of products
Analytical Prep-RP-HPLC: New Method Dramatically Improves Sample Loadability
 In this post, we’ll look at a unique Prep-RP-HPLC methoddeveloped at Neuland Labs to achieve 10-fold increases in sample loadability.
In this post, we’ll look at a unique Prep-RP-HPLC methoddeveloped at Neuland Labs to achieve 10-fold increases in sample loadability.
Despite advances in technology, there are only two ways to increase the amount of sample that can be purified bypreparative reversed phase high performance liquid chromatography – or Prep-HP-LC – in a single run.
Traditionally, you would use a bigger column, extending the stationary phase. The second way is to use displacement chromatography. While labor-intensive, displacement chromatography uses the stationary phase more effectively.
What RP-HPLC is Used For
In science labs, reversed phase high performance liquid chromatography (RP-HPLC) is used to analyze, characterize, separate, purify, and isolate small organic molecules, natural products, and biologically active molecules such as polypeptides, proteins and nucleotides. In pharma, analytical RP-HPLC is employed specifically to release and characterize raw materials, intermediates and active pharmaceutical ingredients (APIs). Likewise, preparative RP-HPLC is used to commercially produce peptide APIs, along with most other complex APIs that cannot be crystallized.
How the New RP-HPLC Method Works
The new method developed by Neuland uses C-18/C-8 derivatized silica, coated with a hydrophobic quaternary ammonium salt or quaternary phosphonium salt. It increases 7- to 12-fold the sample loading of the crude mixture of organic compounds including synthetic crude peptides. What causes such dramatic results is the additional surrogate stationary phase characteristic of the C-18/C-8 bound quaternary salt.
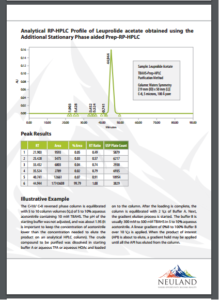
Preparative RP-HPLC in the elution mode is limited by the loading capacity of the analyte. In this mode, however, synthetic peptides typically have a loading capacity of 1-2 mgs per mL of packed column volume (viz., 0.1% to 0.2% with respect to total column volume). Table 1 (found on page 2 of PDF provided at web link) shows how Prep-RP-HPLC aided by a surrogate stationary phase performed compared to standard Prep-RP-HPLC using Leuprolide as an example, and tetra-n-butylammonium hydrogen sulphate (TBAHS) as ASP/SSP.
Results of Neuland’s Prep HPLC Technique
The purified product (Leuprolide) output of the standard Prep-RP-HPLC is 2.45 mg/mL of column volume. In contrast, the purified product output of the Prep-RP-HPLC aided by a surrogate stationary phase is 29.6 mg/mL of the column volume (see Table 1, entry 2, on page 2 at this link) and 16.3 mg/mL of column volume (see Table 1, entry 3, on page 2 at this link). These results demonstrated that the process described enables loadings of 7-12 times the capacity of conventional Prep-RP-HPLC.
To learn more about this novel Prep-RP-HPLC method, view the complete pdf here.
Evaluating Impurities in Drugs (Part I of III)
In Part I of a three-part article, the authors discuss what constitutes an impurity and the potential sources of impurities in APIs and finished drug products.
Intermediates
Organic compounds formed during the synthesis of APIs are termed as intermediates. The compound in the synthetic chain before the production of the final desired compound is called the penultimate intermediate.
Impurities due to rearrangement. Developing practical synthetic routes to render high-yield products in shorter stages or in a one- or two-pot reaction generally involves formation of rearranged intermediates that ultimately give the required final product.
As an example, the cyclization of bromonitrostyrene in the API ropinirole involves the rearrangement of the intermediate cyclic ion to give the indole ring with the formation of hydroxamic ester and chlorooxime acetate as impurities.Impurities due to in situ reactions. Advances in synthetic chemistry have enabled a number of stages in a reaction to be carried out in just one or two pots without the need to isolate intermediates. The downside of such reactions is the unexpected and numerous impurities that form because intermediates and reagents are not isolated.

Figure 3: Linezolid (e.g., oxazolidinones class) and pemetrexed disodium tautomer impurity. EP is the European Pharmacopoeia. RRT is relative retention time.
For example, 4-phenyl butanol is a key raw material for the synthesis of salmeterol Intermediates 1 and 2 (see Figure 2). Intermediate 1 reacts with 4-phenyl butanol in the presence of sodium hydride and toluene to yield Compound 1, which is a nonreactive impurity in further stages. Intermediate 2 reacts with the trace amounts of Intermediate 1 and in the same conditions react to form Compound 2 (see Figure 2).As an example, the alkylation of the key starting material (S)-2-amino butyramide for the API levetiracetam with chlorobutyrylchloride using potassium hydroxide in the presence of tetra-n-butylammonium bromide gives an intermediate that eventually cyclized into levetiracetam. This intermediate, however, is present in the final product as an USP impurity A.Nonreactive intermediates. Nonreactive intermediates are impurities formed in some intermediate stage by the reaction of reagents used in the next stages due to carryover. Such impurities remain nonactive in the later stages.
Reactive intermediates. Reactive intermediates, as the name implies, are byproducts or impurities resulting from the intermediate stages of the reaction that have the potential to react with the reagents or catalysts used in later stages. They are carried forwarded in every stage up to the final API as a reactive intermediate.
During the process development of salmeterol, an unknown impurity was detected at 2.08 RRT at a level of 0.11% and later identified after isolation to be Compound 3 (see Figure 2). The impurity formed in the final API due to presence of N-benzyl-6-(4-cyclohexylbutoxy)hexan-1-amine in Intermediate 2 leads to the salmeterol cyclohexyl impurity (12).
The reactive intermediate, N-benzyl-4-phenylbutan-1-amine is present in Intermediate 2 (see Figure 2). It is formed by the reaction of 4-phenyl butanol with benzyl amine and competes in all reaction stages with Intermediate 2 to form Compound 4 (see Figure 2).
A main challenges faced in developing the olefination route of the API aprepitant was a subsequent reaction of the vinyl ether intermediate with dimethyltitanocene to form an ethyl impurity (13).
Bis-compound impurities. The formation of new or unknown impurities can occur when scaling up a process, even with successful runs at a smaller scale. Examining the molecular weight of such impurities often reveals the compound is exactly double the weight of that being formed in that reaction step. Such dimeric derivatives are called bis-compound impurities. Two bis-compound impurities were formed in the intermediate and final stages in the synthesis of linezolid, to be discussed in Part III of this article.
|
Byproducts
In synthetic organic chemistry, getting a single end product, 100% pure, seldom occurs because of the change into byproducts, which can be formed through a variety of side reactions, such as incomplete reactions, overreactions, isomerization, or unwanted reactions between starting materials, intermediates, chemical reagents, or catalysts. For example, in the bulk production of paracetamol, diacetylated paracetamol may form as a byproduct (14).
In the Claisen rearrangement of the aryl propargyl ether in diethylaniline at elevated temperatures, formation of the desired chroman product is accompanied by the generation of a furan byproduct in success sively increasing amounts (15).

igure 4. Propionaldehyde with malanonitrile reactions. CAS refers to Chemical Abstracts Service, No. is number, and NA is not available. Conditions: (a) with piperidine in pyridine, heating (Ref. 27); (b) with piperidine in pyridine, heating, cyclization (Ref. 28); (c) with piperidine, 1,4-dioxane (Ref. 29–30); (d) With [C4DABCO][BF4] in water, Time = 0.0166667 h, T = 20 °C, Knoevenagel condensation or with aluminum oxide in dichloromethane, T= 20 °C, Knoevenagel condensation aldol-condensation (Ref. 31–33).; and with morpholine in ethanol, T = 20 °C, Knoevenagel condensation (Ref. 34–37).

Figure 5: Reaction scheme of olanzapine impurities. DMF is dimethylformamide. TEA is triethylamine. Addn is addition. RT is room temperature. CAS is Chemical Abstracts Service, No. is number, and NA is not available.
The reaction of propionaldehyde with malononitrile and sulfur resulted in formation of two unknown impurities up to 7%, which were isolated and confirmed by 1 H NMR (nuclear magentic resonance spectroscopy), correlation spectroscopy, nuclear Overhauser effect spectroscopy, and single X-ray crystallography to be Impurity 1 (see Figures 4 and 5). These impurities are further found to react with 2-fluoro nitrobenzene to give next-stage impurities and which are controlled by purification in the respective stages.In the ropinirole synthesis, a somewhat similar case is observed in the final step. The reaction between the ropinirole precursor 4-(2-bromoethyl)-13-dihydro-2H-indol-2-one and di-n-propyl amine in water produces ropinirole in modest yield (57%), together with styrene as the major byproduct (38%) (16).In another example, thiophenes are important heterocyclic compounds that are widely used as building blocks in many agrochemicals and pharmaceuticals (17). The synthesis of 2-amino-5-methylthiopene-3-carbonitrile is achieved by reacting a mixture of sulfur, propionaldehyde, malononitrile, and dimethylformamide using triethylamine (18–26).
mpurity 1 (see Figure 5) is a novel tricarbonitrile bicyclic compound, and as of the writing of this article, it is not known in the literature. Prediction of cLogP is 0.65, drug linkness is 4.04, and the drug score is 0.45 as determined by OSIRIS Property Explorer, software used to calculate various drug-relevant properties of chemical structures. Structure–activity relationship, quantitative structure–activity relationship, and drug design with other modified organic/inorganic hetrocyclic moieties could give some biological activity. The molecular designing of Impurity 1 for specific and unspecific purposes (e.g., DNA-binding, enzyme inhibition, anticancer efficacy) is based on the knowledge of molecular properties, such as the activity of functional groups, molecular geometry, and electronic structure, and on information cataloged on analogous molecules. The compound 2,6-diamino-7-ethyl-8-methylbicyclo[2.2.2]octa-2,5-diene-1,3,5-tricarbonitrile could be coupled with an active or nonactive peptide to check the biological activity as a prodrug or drug. The potential therapeutic and prophylactic activities of antimalarials, antimitotics, and antitumor agents could also be performed. This bicyclic compound may be used alone as a single agent or in combination with any organic or inorganic salts in chemotherapy or in combination with other chemotherapeutic agents after in vivo and in vitro testing
.
|
Transformation products
Transformation products deal with theorized and nontheorized products produced in a reaction. They can be synthetic derivatives of byproducts and are closely related to byproducts.

Figure 6: Chloro impurity-formation scheme of salmeterol. HPLC is high-performance liquid chromatography. MDC is methylenedichloride; AlCl3 is aluminum chloride.
The term interaction product deals with the interaction of two or more intermediates/compounds with various chemicals, intentionally or unintentionally. An interaction product is slightly more comprehensive than byproducts and transformation products. Two types of interaction products that are commonly encountered are drug substance–excipient interactions and drug substance–container/closure interactions.A reaction where transformation products occur is the formation of chloro acetyl derivative of salicylaldehyde during the acylation reaction of salicylaldehyde with bromo acetyl bromide using methylenedichloride (MDC) and aluminum chloride (AlCl3). Mechanistically, the formation of chloroacetyl derivative using bromoacetyl bromide could not be expected, but hypothetically, it could occur as a transformation reaction due to halogen exchange. During Friedel–Craft acylation with Lewis acid AlCl3 in methylene dichloride, the Lewis acid forms an ionized complex [Cl–AlCl2–Br]–, which eventually undergoes halogen exchange with the bromo acylium ion to yield the chloro acetyl derivative. Formation of this impurity in reaction is as high as 7–20%, which is an uncontrolled impurity in the manufacturing process. Nevertheless, this impurity would not affect the purity of the final drug substance because the reaction of the transformed impurity with 2 (see Figure 6, Part I) forms the desired product, salmeterol. The presence of the chloro impurity also has been confirmed by experiment (see Figure 6, Part II).Interaction products
Related products
The term related products means that the impurity has similar structure as that of the drug substance and may exhibit similar biological activity. This structural similarity by itself, however, does not provide any guarantee of similar activity. An example of a related product is 8-fluoro olanzapine.
Degradation products
Impurities formed by decomposition or degradation of the end product during manufacturing of the bulk drug are called degradation products. The term also includes degradation products resulting from storage, formulation, or aging. Parts II and III of this article will discuss the types and sources of the degradation products in further detail.
Tautomer impurities
Tautomers are readily interconvertible constitutional isomers that coexist in equilibrium. For APIs or drug molecules that exhibit tautomerism, there has been a confusion in identifying the two tautomeric forms. If one tautomer is thermodynamically stable and is the major form, the other tautomer should be considered as an impurity or simply termed as a tautomer of the API or drug molecule. To the best of the authors’ knowledge, there has been no literature relating to the isolation, synthesis, or characterization of a tautomeric impurity(-ies) from the final API.
Linezolid is an treatment for nosocomial infections involving gram-positive bacteria. Oxazolidinones possess a unique mechanism of bacterial protein synthesis inhibition (38–39). Linezolid has an N-acetyl group (–NH–CO–CH3) due to that lactam–lactim tautomerism, which may occur during the synthesis but also may be stable. An effective analytical method needs to be developed to identify both tautomers.
A key starting raw material of pemetrexed disodium 2,4-diamino-6-hydroxy-pyrimidine shows the keto-enol form occurring in different ratios and which will be converted to the final drug using a known synthesis (see Figure 3).
Tautomers vary in their kinetic and thermodynamic stability, thereby making it difficult to determine whether they could be separated, isolated, or analyzed. Keeping this in mind, the use of the term impurity for tautomers in a final API/drug moiety presumably will be an important discussion in near future.
Conclusion
Part I of article highlights the origination and classification of impurities and provides a perspective on impurities in drug substances and drug products. The impurity profile of a drug substance is on increasing importance for ensuring the quality of drug products. Whatever the class of impurity, its identification and adequate control is a tremendous challenge for process-development chemists. Because no two drugs are alike, neither are two development pathways. Each drug candidate poses a different challenge in terms of impurities, and establishing efficient ways for the isolation and control of impurities is a key task in process development.
References
1. FDA, Guideline for Submitting Supportive Documentation in Drug Applications for the Manufacture of Drug Substances (Rockville, MD, Feb. 1987).
2. ICH, Q7 Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients, Step 5 (Nov. 2000).
3. FDA, Draft Guidance for Industry: Drug Substance: Chemistry Manufacturing and Controls Information (Rockville, MD, Jan. 2004).
4. T. Cupps et al., Pharm. Technol. 27 (2), 34–52 (2003).
5. M. Johnson, Med. Res. Rev. 15 (3), 225–257 (1995).
6. A.T. Nials et al., Am. Rev. Resp. Dis. 149, A481 (1995).
7. Y Kawakami et al., Eur. J. Med. Chem. 31 ( 9), 683–692 (1996).
8. N.O. Mahmoodi and M. Jazayri, Syn. Comm. 31 (10), 1467–1476 (2001).
9. M. Islam et al., Acta Poloniae Pharm. Drug Res., 65 (4) 441–447 (2008).
10. K.T. Chapman et al., Bioorg. Med. Chem. Lett. 6 (7), 803–806 (1996).
11. A.A. Siddiqui et al., Bioorg. Med. Chem. Lett. 21 (3), 1023–1026 (2011).
12. B. Venkatasubbaiah et al., Scientia Pharm. 77, 579–587 (2009) .
13. J.J. Hale et al., J. Med. Chem. 41 (1), 4607–4614 (1998).
14. K.M. Alsante, Amer. Pharm. Rev. 4 (1) 70–78 (2001).
15. J. Zsindely et al., Helv. Chim. Acta 51, 1510 (1968).
16. J.D. Hayler et al., Org. Process Res. Dev. 2 (1), 3–9 (1998).
17. J. Swanston, “Thiophene” in Ullmann’s Encyclopedia of Industrial Chemistry, (Wiley-VCH, Weinheim, Germany, 2006).
18. Tel-Aviv University, “Novel Psychotropic Agents Having Glutamate NMDA Activity,” WIPO Patent WO2008/50341, May 2008.
19. Watson Pharmaceuticals, “2-Methyl-thieno-benzodiazepine Process,” WIPO Patent WO2004/94390, Nov. 2004.
20. Shastri et al., “Process for Producing Pure Form of 2-Methyl-4-(4-Methyl-1-Piperazinyl)-10H-Thieno[2,3-b] [1,5]Benzodiazepine,” US Patent 2009/5556, Jan. 2009.
21. Eli Lilly, “Process for Preparing 2-Methyl-thieno-benzodiazepine” US Patent 6008216, Dec. 1999.
22. Lilly Industries, “2-Methyl-thieno-benzodiazepine,” US Patent 5229382, July 1993.
23. Eli Lilly, “2-Methyl-thieno-benzodiazepine,” US Patent 5605897, Feb. 1997.
24. X He et al., J. Pharm. Sci. 90 (3) 371–388 (2001).
25. V.P. Shevchenko, Russian J. Bioorg. Chem. 31 (4), 378–382 (2005).
26. V.P. Shevchenko, Bioorganicheskaya Khimiya 31 (4) 420–424 (2005).
27. J.C. Dunham et al., Synthesis, 4, 680–686 (2006).
28. A.H. Elgandour et al., Indian J. Chem. Sec. B: 36 (1) 79–82 (1997).
29. R. Mariella and A. Roth, J. Org. Chem. 22 (9), 1130 (1957).
30. Hart and Freeman, Chemistry and Industry, p. 332 (1963).
31. Da-Zhen Xu et al., Green Chem. 12 (3) 514–517 (2010).
32. H.C. Brown and M.V. Rangaishenvi, J. Heterocycl. Chem. 27 (1), 1–12 (1990).
33. S. Fioravanati, Synlett. (6), 1083–1085 (2004).
34. V.D. Dayachenko, J. Gen. Chem. 74 (7), 1135–1136 (2004).
35. Zhurnal Obshchei Khimii 74 (7), 1227–1228 (2004).
36. V.D. Dayachenko and A.N. Chernega, Russian J. Org. Chem. 42 (4), 567–576 (2006).
37. Zhurnal Organicheskoi Khimii 42 (4), 585–593 (2006).
38. D.L.K. Marotti et al., AntiMicrob. Agents Chemother. 41 (10), 2132–2136 (1997).
39. E.Z. Gray et al., Expert Opin. Investig. Drugs 6 (2), 151–158 (1997).
Evaluating Impurities in Drugs (Part 2 of III)
Figure 1: Reaction scheme for different process approaches for pemetrexed sodium impurities, respectively labeled as 1, 2, 3, and 4 (Refs. 57–60). Ph. Eur. is European Pharmacopoeia.
Impurities due to the piperazine ring
The piperazine moiety is present in the chemical structure of more than 200 drugs. The biotransformation of the piperazine ring involves several well-known metabolic reactions, including N-oxidation, hydroxylation, N-dealkylation, and ring cleavages toN-substituted as well as N,N‘-disubstituted ethylenediamines. In addition, several unexpected metabolic pathways have been reported for the piperazine ring: N-glucuronidation, N-sulfonation, formation of carbamoyl glucuronide, and glutathione adducts (61). Some compounds containing the piperazine ring indicate that the ring is normally metabolically stable when both nitrogen atoms are substituted with groups larger than ethyl.
The lack of partial degradation of the piperazine ring to form ethylenediamine in olanzapine (2-methyl-4-(4-methyl-1-piperazinyl)10H-thieno[2,3-b][1,5]benzodiazepine) is slightly surprising. Some major metabolites were reported in humans plasma and urine, such as 4′-N-glucuronide and 4′-N-glucuronide (61, 62). Several other metabolites also were reported in mice, rats, monkeys, and dog urine (63). The ethylenediamine impurity, however, is not reported as a metabolite and a process impurity (see Figure 2).When one of the nitrogen atoms is substituted by hydrogen on the piperazine ring, whether its methyl or ethyl, ethylenediamine formation is normally observed. An example is levofloxacin, S-(-)-9-fluoro-2,3-dihydro-3-methyl-10-(4-methyl-1-piperazinyl)-7-oxo-7H-pyrido[1,2,3-de][1,4]benzoxazine-6-carboxylic acid, which is the (S)-isomer of ofloxacin. In levofloxacin, the piperazine nitrogen atom is substituted with methyl due to several photodegradation impurities (see P 2 to P 10, Figure 3) (64–67). Some process impurities also are observed (see Figure 3). If the levofloxacin process involved methylenedichloride as a solvent, a chloro methyl impurity may form, and after isolation of the final product, the same impurity may convert to a di-quaternary cyclic piperazine impurity.Additionally, when the ciprofloxacin (1-cyclopropyl-6-fluoro-1,4-dihydro-4-oxo-7-(piperazin-1-yl) quinoline-3-carboxylic acid) nitrogen atom is substituted by hydrogen on the piperazine ring, several metabolites and process impurities are formed (see Figure 3) (68–74). When nitrogen is substituted with hydrogen during the reaction, two dimer impurities (F-F dimer ciprofloxacin and F-Cl dimer ciprofloxacin) also are observed (75).

Figure 2: The piperazine ring and metabolite impurities of olanzanpine. Ph. Eur. is European Pharmacopoeia. USP is US Pharmacopeia.
Figure 3: The piperazine ring and metabolite impurities of levofloxacin and ciprofloxacin. Ph. Eur. is European Pharmacopoeia and USP is US Pharmacopeia. CAS No. refers to Chemical Abstracts Service (CAS) number.
Genotoxic impurities
There was no specific document on control of genotoxic impurities before 2000. ICH guidelines made passing references to compounds of unusual toxicity. Genotoxic impurities are chemical compounds that may be mutagenic and could potentially damage DNA (76). Non-monoalkylated agents are classified as genotoxic due to the nature of the functional groups they possess and also of related aniline derivatives. Additionally, salt-forming steps can introduce genotoxic impurities. Some examples include formation of methyl chloride as a side reaction of hydrochloric acid in methanol or esters of methanesulfonic acid as byproducts from the methanesulfonic acid salt-formation step in alcohol-based solvents (77, 78).
EMA issued guidelines on the threshold of toxicological concern (TTC) that recommended limits for exposure to potential genotoxic impurities to be 1.5 mcg per day for commercially approved drugs (79). As per the guidelines, testing will be required for all potential impurities from an API’s synthetic route containing structural elements that are the cause of concern for genotoxicity potential using the well-established Salmonelle Ames test. The Ames test is a screening test that is used to help identify chemicals that affect the structure of DNA. The test exposes Salmonella bacteria to chemicals and looks for changes in the way bacteria grow. These changes result from mutations that occur when the structure of DNA is altered in certain places and the micronuclei test for mutagenicity (80, 81). Recommended qualification thresholds based on the maximum daily dose for drug substances and for drug products are provided in ICH Q3A and ICH Q3b (7, 8). The TTC data set was conducted from the perspective of an organic chemist who develops process technology for APIs (82). As part of the EMA guidance, API process designers are instructed to avoid all possible situations that could lead to the presence of impurities possessing genotoxic potential at any level in APIs.
During the establishing of the control mechanism, other factors, such as reactivity, solubility, and volatility, should be considered. Action should not be based only on the presence of alerting structures. It is important to make evaluations on a case-by-case basis, and precedence data should be considered, such as the stage of impurity formation, reactivity and carryover to the API, the intake of other routes, Ames test results, and data of closely related structures.During process development, a genotoxic impurity may be introduced as a starting material, reagent, intermediate, catalyst, byproduct, isomer, or degradation product. (83). Alkyl halides used as reagents in synthesis are genotoxins (84). The same also was generated during chemical synthesis when a salt counter ion (e.g., hydrogen halide) of a drug substance reacts with alcohols when used as a solvent media.
The genotoxins ethyl chloride, methyl chloride, and isopropyl chloride were generated during the preparation of the hydrochloride salts of ethanol, methanol, and isopropyl alcohol (ICH listed solvents), respectively, at lower temperature (< 5 °C) as the key parameter of these impurities. In alcohol solvents, when HCl was 37% aqueous HCl or gas, it creates the maximum chance to form these alkyl halide impurities at trace levels. These impurities are detectable in GC at ppm level. Methane sulfonic acid (mesylate), benzene sulfonic acid (besylate) and p-toluenesulfonic acid (tosylate) are commonly used as counter ions to form API salts (85–87). Interactions of these acids with residual alcohols may lead to the generation of genotoxic impurities. Alkyl methane sulfonates, alkyl benzene sulfonates, and alkyl para-toluene sulfonates may combine with imatinib mesylate, amlodipine besylate, and denagliptin tosylate, respectively (88, 89).
The emphasis on genotoxic impurities is increasing, which creates challenges for both synthetic and analytical chemists, to develop sensitive and efficient methods to detect impurities at low levels (i.e., below TTC < 1.5 mcg/per day), which sometimes is not feasible and which increases the time and cost of drug development.
 Linezolid (S)-N-[[3-[3-fluoro-4-(4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]-acetamide, has genotoxic structural alerts and represents a new class of antibiotics, oxazolidinones. Forced-degradation studies are an important part of the drug-development process and are used increasingly in testing new molecules. These studies may give different impurities that may not be formed during process optimization and manufacturing validation, but these impurities must be controlled as per ICH guidelines. The authors have observed two impurities during a forced-degradation study in peroxide and alkaline conditions, Compounds 7, 8, and 9 (see Figure 4), which are structural alerts for genotoxicity, and which should be controlled so that the exposure to it is less than 1.5 mcg/day based on the maximum daily dose of the linezolid.Linezolid’s key starting material (A) shows genotoxicity alert and it contains five other intermediates, Compounds 1, 2, 3, 4, and 5 (see Figure 4). Compound A converts to the final drug, and it contains, Mesyl Impurity 6, Amine Impurity 12, Des Fluoro Impurity 13, Chloro Impurity 14, and O-Acetyl Impurity 15; these are the process impurities and have genotoxicity alert (49). During human studies, from the total amount of linezolid administered, only 30% was eliminated through the kidneys. Its major part was metabolized by oxidation of its morpholine ring, which resulted in the formation of two metabolites (see Figure 4): amino ethoxy acetic acid metabolite and hydroxy ethyl glycine metabolite (i.e., a major urinary metabolite) (90–92).
Linezolid (S)-N-[[3-[3-fluoro-4-(4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]-acetamide, has genotoxic structural alerts and represents a new class of antibiotics, oxazolidinones. Forced-degradation studies are an important part of the drug-development process and are used increasingly in testing new molecules. These studies may give different impurities that may not be formed during process optimization and manufacturing validation, but these impurities must be controlled as per ICH guidelines. The authors have observed two impurities during a forced-degradation study in peroxide and alkaline conditions, Compounds 7, 8, and 9 (see Figure 4), which are structural alerts for genotoxicity, and which should be controlled so that the exposure to it is less than 1.5 mcg/day based on the maximum daily dose of the linezolid.Linezolid’s key starting material (A) shows genotoxicity alert and it contains five other intermediates, Compounds 1, 2, 3, 4, and 5 (see Figure 4). Compound A converts to the final drug, and it contains, Mesyl Impurity 6, Amine Impurity 12, Des Fluoro Impurity 13, Chloro Impurity 14, and O-Acetyl Impurity 15; these are the process impurities and have genotoxicity alert (49). During human studies, from the total amount of linezolid administered, only 30% was eliminated through the kidneys. Its major part was metabolized by oxidation of its morpholine ring, which resulted in the formation of two metabolites (see Figure 4): amino ethoxy acetic acid metabolite and hydroxy ethyl glycine metabolite (i.e., a major urinary metabolite) (90–92).
Figure 4: Process, genotoxic, and metabolite impurities of linezolid.
Conclusion
Part II of this article examined impurities that are associated with drug molecules having one or more chiral centers, APIs existing in various crystalline forms, drug substances with the piepererazine moiety, and APIs developed by new processes. Part II also looked into the extended application of the TTC to pharmaceuticals. To guarantee the quality and safety of pharmaceuticals during drug development, a quality concept has been proposed that adapts the ICH guidelines and which is focused on qualified impurity profiles.
References
1. FDA, Guidance for Industry—ANDAs: Impurities in Drug Products (Rockville, MD, Aug. 2005).
2. FDA, Guidance for Industry—ANDAs: Impurities in Drug Substances (Rockville, MD, Jan. 2005).
3. S. Görög, Identification and Determination of Impurities in Drugs (Elsevier Science, Amsterdam, 2000).4. S. Ahuja, Impurities Evaluation of Pharmaceuticals (Marcel Dekker, New York, 1998).
5. S. Hovorka and C. Schöneich, J. Pharm.Sci. 90 (3), 253–269 (2001).
6. J. Roy, AAAPS PharmSciTech 3 (2), 1–8 (2002).
7. ICH, Q3A(R) Impurities in New Drug Substances (Feb. 2003).
8. ICH, Q3B(R) Impurities in Drug Products (Nov. 2003).
9. ICH, Q3C (R5) Impurities: Guideline for Residual Solvents (March 2011).
10. ICH, Q1A(R2) Stability Testing of New Drug Substances and Products (Nov. 2003).
11. K. R. Wadekar et al., Pharm. Technol. 36 (2), 46–51 (2012).
12. B.C. Allen, K.S Crump, and A.M. Shipp, Risk Anal.8 (4), 531–544 (1988).
13. S.W. Baertschi and D.W. Reynolds, “Introduction” in Pharmaceutical Stress Testing: Predicting Drug Degradation, J. Swarbick, Ed. (Taylor & Francis, New York, 2005), pp. 4–8.
14. S. Ahuja, Chiral Separations by Chromatography (Oxford University Press, New York, 2000).
15. S. Ahuja, Chiral Separations by Liquid Chromatography, ACS Symposium Series 471 (American Chemical Society, Washington, DC, 1991).
16. J. Trofast et al., Chirality 3 (6), 443–450 (1991).
17. B. Waldeck, Chirality 5 (5) 350–355 (1993).
18. L. Gillespie et al., Circulation 25, 281–291 (1962).
19. H. Kubota et al., Chem. Pharm. Bull. 40, 1619–1622 (1992).
20. R.B. Carter, J. Pharmacol. Exp. Ther. 234 (2), 299–306 (1985).
21. Chiral Agonists of Histamine in Fornitier in Histamine Research (Oxford, 1985), pp.39-46.
22. M.E. Goldman et al., J. Mol. Pharmacol. 25 (1), 18–23 (1984).
23. W.M. Welch et al., J. Med. Chem. 27 (11), 1508–1515 (1984).
24. B.K. Koe et al., J. Pharmacol. Exp. Ther. 226 (3), 686–700 (1983).
25. T. de Boer et al., Chromatogr. 26 (2), 156–165 (2012).
26. TGA, Australian Public Assessment Report for Asenapine (Woden, Australia, April 2011).
27. S.G. Allenmark, Chromatographic Enantioseparation: Methods and Applications(Ellis Horwood, Chichester, 1991).
28. G. Gubitz, Chromatographia 30, 555–564 (1990).
29. D.E. Drayer, Clin. Pharmacol. Ther. 40 (2), 125–133 (1986). |
30. F.J. Jamali, J. Pharm. Sci. 78 (9), 695–715 (1989).
31. “FDA’s Policy Statement for the Development of New Stereoisomer Drugs,” Chirality4 (5), 338–340 (1992).
32. H. Wilson et al., J. Pharm. Biomed. Anal. 11 (11–12), 1167–1173 (1993).
33. A.G. Rauws, Chirality 6 (2), 72–75 (1994).34. M. Gross et al., Drug Inf. J. 27 (2), 53–457 (1993).
35. W.L. Heydorn, Pharm. News 2, 19–21 (1995).
36. ICH, Q6A Document, Specification for New Drug Substances and Products: Chemical Substances, Step 2 version (1992).
37. J. Haleblian et al., J. Pharm. Sci. 58 (8) 911–929 (1969).
38. G.A. Stephenson et al., Adv. Drug. Deliv. Rev. 48, 67–90 (2001).
39. ICH, Q6A Specification: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances (1999).
40. G.G. Z. Zhang et al., Adv. Drug Deliv. Rev. 56 (3), 371–390 (2004).
41. S.R. Vippagunta et al., Adv. Drug Deliv. Rev. 48 (1), 3–26 (2001).
42. P.B. Molinoff, The Pharmacological Basis of Theraputics (McGraw Hill, New York, 1996), pp. 399–430.
43. S.M. Reutzel-Edens, Cryst. Growth Des. 3 (6) 897–907 (2003).
44. Eli Lilly, “Olanzapine Polymorph Crystal Form,”US Patent 5,736,541 (April 1998).
45. H.Y. Tong et al., Pharm. Res. 18 (6), 852–858 (2001).
46. H.H. Tong et al., Pharm.Res. 20 (9), 1423–1429 (2003).
47. D. Clemett et al., Drugs 59 (4), 815–827 (2000).
48. Teva Pharmaceutical Industries, “Isolated Bis-Linezolid, Preparation Thereof and Its Use as a Reference Standard, WO 2006/091848 A2, Aug. 2006.
49. Neuland Laboratories, “A Process for the Preparation of (5S)-(N)-[[3-[3-fluoro-4-4(4-morpholinly)phenyl-2-oxo-5-oxazolidinyl]methyl] acetamide,” WO2010084514A2, July 2010.
50. Symed Labs, “Novel Process for the Preparation of Linezolid and Related Compounds,” US Patent 2007/0032472, Feb, 2007.
51. B. A. Peralman, “Process to Produce Oxazolidiones,” US Patent 2002/0095054, July 2002.
52. Pfizer, “Process for Preparing Linezolid,” WO 2007/116284, Oct. 2007.
5 3. Pharmacia & Upjohn, “Process to Prepare Oxazolidinones,” US Patent 5837870, Nov. 1998.
54. Jubilant Life Sciences, “Process for the Preparation of Linezolid,” WO 2011/114241, Sept. 2011.
55. Pharmacia and Upjohn, “Substituted Oxazine and Thiazine Oxazolidinone Antimicrobials,” US Patent 5688792, Nov. 1997.
56. D.C.M Chan et al., Curr. Med. Chem. 13 (4), 377–398 (2006).
57. Fortress Metro Hainan Tianyuan Pharmaceutical Technology Co., Ltd., “Nitro Compounds and the Preparation of Pemetrexed CN1827604A, Sept. 2006.
58. Wu Torrent, “Pemetrexed Intermediates and Preparation Methods, CN101085775A, Dec. 2007.
59. Dr. Reddy’s Laboratories, “Process for Preparing Pemetrexed, WO 2011/019986 A2, Feb. 2011.
60. D.P. Kjell et al., Org. Proc. Res. Dev. 9 (6), 738–742, 2005.
61. K. Kassahun et al., Drug Metab. Dispos. 25 (1), 81–93 (1997).
62. K. Kassahun et al., Drug Metab. Dispos. 26 (9), 848–855 (1998).
63. E. Mattiuz et al., Drug Metab. Dispos. 25 (1), 573–583(1997).64. M. Lalitha Devi et al., J. Pharm. Biomed. Anal. 50 (5), 710–717 (2009).
65. Y. Yoshida et al., Arzneimittelforschung 43 (5), 601–606 (1993).
66. J. Sunderlanda et al., J. Antimicrob. Chemother. 47 (3), 271–275 (2001).
67. A. V. Polishchuk et al., High Energy Chemistry 42 (6), 459–463 (2008).
68. W. Gau, et al., Arzneimittel Forschung 36 (10), 1545–1549 (1986).
69. P. Mojaverian et al., J. Pharm. Biomed. Anal. 16 (3), 439–445 (1997).
70. A. Taicheng et al., Applied Catalysis B: Environmental 94 (3–4), 288–294, 2010.
71. M. Mella et al., Helvetica Chimica Acta 84 (8), 2508–2519 (2001).
72. K.A. Thabaj et al., Polyhedron 26 (17), 4877–4885 (2007).
73. T.G. Vasconcelos et al., Chemosphere 76 (4), 487–493 (2009).
74. K. Torniainen et al., J. Pharm. Biomed. Anal. 15 (7), 887–894 (1997).
75. K. Tovarna Zdravil, “Process for Preparing Purified Ciprofloxacin,” WO 2005/075430, Aug. 2005.
76. R. J. Islam M et al., J. Pharm. Sci. 90 (5), 541–544 (2001).
77. H.V Hogerzeil et al., British Medical Journal 304, 210–214 (1992).
78. R. J. Bhuiyan K et al., Indian Drugs 34 (11), 634-636 (1997).
79. EMA, Guideline on the Limits of Genotoxic Impurities (London, June, 2006).
80. B.N. Ames and L.S. Gold, Proc. Nat. Acad. Sci. USA 87 (19), 7772–7776 (1990).
81. B.N. Ames and L.S. Gold, Proc. Nat. Acad. Sci. USA 87 (19), 7777- 7781 (1990).
82. M.A. Cheeseman et al., Food Chain Toxicol. 37, 387–412 (1999).
83. D.A. Pierson et al., Org. Proc. Res. Dev. 13 (2), 285–291 (2009).
84. D.P. Elder. et al., J. Pharm. Biomed. Anal. 48 (3), 497–507 (2008).
85. D.P. Elder. et al., J. Pharm. Biomed. Anal. 46 (1), 1–8 (2008).
86. D.P. Elder. et al., J. Pharm. Sci. 99 (7,) 2948-2961 (2010).
87. G.E. Taylor. et al., J. Chromatogr A 1119 (1–2), 231–237 (2006).
88. K. Ramakrishna. et al., J. Pharm. Biomed. Anal. 46 (4), 780–783 (2008).
89. N.V.V.S.S. Raman. et al., J. Pharm. Biomed. Anal. 48 (1), 227–230 (2008).
90. J. G. Slatter et al., Drug Metab. Dispos. 29 (8), 1136–1145 (2001).
91. N. Plock et al., Drug Metab. Dispos. l35 (10), 1816–1823 (2007).
92. Wynalda et al., Drug Metab. Dispos. 28 (9), 1014–1017 ( 2000). |
Evaluating Impurities in Drugs (Part 3 of III)
Controlling and monitoring impurities in APIs and finished drug products is a crucial issue in drug development and manufacturing. Part I of this article, published in the February 2012 issue of Pharmaceutical Technology, discussed the various types of and sources of impurities with specific case studies (1). Part II, published in the March 2012 issue, examined chiral, polymorphic, and genotoxic impurities (2). In Part III, the authors examine various degradation routes of APIs, impurities arising from API–excipient interaction during formulation, metabolite impurities, various analytical methodologies to measure impurity levels, and ways to control impurities in pharmaceuticals.
Definition of impurity
The term impurity reflects unwanted chemicals that are present in APIs or that develop during formulation or upon aging of the API in the formulated drug product. The presence of such unwanted material, even in small amounts, could affect the efficacy and safety of pharmaceutical products. Several guidelines from the International Conference on Harmonization (ICH) address impurities in new drug substances, drug products, and residual solvents (3–6). As per the ICH guidelines on impurities in new drug products, impurities present below a 0.1% level do not need to be qualified unless the potential impurities are expected to be unusually potent or toxic (5). In all other cases, impurities should be qualified. If the impurities exceed the threshold limits and data are not available to qualify the proposed specification level, studies to obtain such data may be required. Several recent articles describe a designed approach and guidelines for isolation and identification of process-related impurities and degradation products using mass spectrometry, nuclear magnetic resonance (NMR) spectroscopy, high-performance liquid chromatography (HPLC), and Fourier transform infrared (FTIR) spectroscopy for pharmaceutical substances (7–9).
Degradation-related impurities

Figure 1: Degradation of hydrochlorothiazide. (ALL FIGURES ARE COURTESY OF THE AUTHORS) |
Degradation products are compounds produced by decomposition of the material of interest or active ingredient. Several impurities may result because of API degradation or other interaction on storage, so stability studies need to be conducted to ensure drug product safety (10). Hydrochlorothiazide (see Figure 1) is a classical example of a degradation impurity. It has a known degradation pathway through which it degrades to the starting material as disulfonamide in its synthesis.
Degradation products could result from the synthesis itself, storage, formulation of the dosage form, and aging (11). These degradation pathways are further discussed.
Synthesis-related impurities. Impurities in a drug substance or a new chemical entity originate mainly during the synthetic process from raw materials, solvents, intermediates, and byproducts. The raw materials are generally manufactured to much lower purity requirements than a drug substance, and thus, it is easy to understand why they can contain a number of components that can in turn affect the purity of the drug substance.
1-Methyl-3-phenyl piperazine (see Figure 2) is present as an unreacted starting material that competes in all the stages eventually leading to the impurity keto-piperazine derivative of mirtazapine (see Impurity C, Figure 2).

Figure 1: Degradation of hydrochlorothiazide

Figure 2: Reaction scheme for mirtazapine impurity. Ph. Eur is the European Pharmacopoeia. DMF is dimethylformamide. EtOAc is ethyl acetate.
Formulation-relatedimpurities . Several impurities in a drug product or API can arise from interactions with excipients used to formulate the drug product. In the process of formulation, a drug substance is subjected to various conditions that can lead to its degradation or other deleterious reactions. For example, if heat is used for drying or for other reasons, it can facilitate degradation of thermally labile drug substances. Solutions and suspensions are potentially prone to degradation due to hydrolysis or solvolysis. These reactions also can occur in the dosage form at solid state, such as in the case of capsules and tablets, when water or another solvent has been used for granulation.
There are two typical conditions in solid- and solution-state degradation studies. Typical conditions for the API in a solid state might be 80 °C, 75% relative humidity (RH); 60 °C at ambient RH; 40 °C at 75% RH; and light irradiation. Typical conditions for an API in the solution state might be: pH 1–9 in buffered media; with peroxide and/or free-radical initiator; and light irradiation.
Method-related impurities. A known impurity,1-(2,6-dichlorophenyl)indolin-2-one is formed in the diclofenac sodium ampuls. Formation of this impurity depends on the initial pH of the preparation and the conditions of sterilization (i.e., autoclave method, 123 °C ± 2 °C) that enforces the intermolecular cyclic reaction of diclofenac sodium, forming indolineone derivative and sodium hydroxide (16).Figure 3 shows the degradation pathway of ketorolac in the solid and solution states (12–14).Dosage form-related impurities. Impurities related to the dosage form are significant because many times precipitation of the main ingredient requires various factors, such as pH or leaching, to be altered (15). For example, the precipitation of imipramine hydrochloride with sodium bisulfite requires a subsequent pH alteration of lidocaine hydrochloride solution in the presence of 5% dextrose in saline.
Environmental-related impurities . Environmental-related impurities may result from the following:
- Temperature. Many heat-labile compounds, when subjected to extreme temperature, lose their stability. Keeping this in mind, extreme care should be exercised to prevent them from degradation.
- Light (ultraviolet light). Exposure to light results in a photolytic reaction. Several studies reported that ergometrine and ergometrine injections are unstable under tropical condition such as light and heat (17–19).
- Humidity. Humidity is one of the important factors when working with hygroscopic compounds. Humidity can be deleterious to bulk powders and formulated solid dosage forms. Well-know examples are ranitidine and aspirin (19).

Figure 3: Degradation pathway of ketorolac.
Impurities on aging. Generally, a longer stay on the shelf increases the possibility that impurities will occur. Such impurities can be caused by several interactions as further described.
- Interaction among ingredients. Vitamins are highly prone to instability after aging. For example, the presence of nicotinamide containing four vitamins (nicotinamide, pyridoxine, riboflavin, and thiamin) caused the degradation of thiamin to a substandard level during a one-year shelf life (20). Table I lists some examples of interactions among ingredients.
- Hydrolysis. Many drugs are derivatives of carboxylic acids or contain functional groups susceptible to acid–base hydrolysis (e.g., aspirin, atropine, and chloramphenical).
- Oxidation. In pharmaceuticals, the most common form of oxidative decomposition is auto-oxidation through a free-radical chain process. Drugs that are prone to oxidation include methotrexate, adinazolam, catecholamine, conjugated dienes (i.e., vitamin A), and nitroso and nitrite derivatives. Olanzapine is especially prone to oxidative degradation in the presence of oxygen (see Figure 4) (21).
- Photolysis. Photolytic cleavage on aging products occurs with APIs or drug products that are prone to degradation on exposure to UV light. For example, the ophthalmic formulation of ciprofloxacin drops 0.3%, when exposed to UV light and undergoes photolysis to form ethylene diamine, an analog of ciprofloxacin (22).
- Decarboxylation. Carboxylic acid (–COOH) tends to lose carbon dioxide from carboxyl groups when heated. For instance, a photoreaction of a rufloxacin enteric tablet coated with cellulose acetate phthalate and subcoated with calcium carbonate causes hydrolysis of cellulose acetate phthalate. This reaction liberates acetic acid, which on reacting with calcium carbonate, produces carbon dioxide as a byproduct.
- pH. It is well understood that pH, particularly extreme levels of pH, can encourage hydrolysis of the API when ionized in an aqueous solution. This situation necessitates buffer control if such a dosage form is required.
- Packaging materials. Impurities may result from packaging materials (i.e., containers and closures (23).
From a regulatory perspective, forced degradation provides data to support the following (25):Two impurities in olanzapine have been identified as 1 and 2 (see Figure 4) (24). The structures indicate that the two impurities are degradation products resulting from oxidation of the thiophene ring of olanzapine.
- Identification of possible degradants
- Degradation pathways and intrinsic stability of the drug molecule
- Validation of stability for indicating analytical procedures
- Facilitation of the development of analytical methods to evaluate stability
- Understanding the degradation of the API to a rational product.
- Screening for possible formation of potential genotoxins.
Various issues are addressed in regulatory guidance (3–6). Some key issues are:
- Forced degradation is typically carried out using one batch of material.
- Forced-degradation conditions are more severe than accelerated stability testing, such as 50 °C; ≥ 75% RH; light conditions exceeding ICH standards; high and low pH; and oxidation.
- Photostability should be an integral part of forced-degradation study design (10).
- Degradation products that do not form in accelerated or long-term stability may not have to be isolated or have their structure determined.
- Mass balance should be considered.
Various issues are not addressed in regulatory guidance (3-6). Some key issues not addressed are:
- Exact experimental conditions (temperatures, duration, and extent of degradation)
- Experimental design (left to the applicant’s discretion).

Figure 4: Olanzapine impurities due to air, heating, and formulation
Metabolite impurities
Metabolite impurities are byproducts formed in the body after a drug substance is ingested. During metabolism, the API and drug product in the body are exposed to various enzymes, from which metabolite impurities can be formed (26–34). Drug metabolism is traditionally divided into two phases: metabolic (i.e., hepatic) clearance and the Phase I and Phase II process. The division is based on the observation that a drug substance first undergoes oxidative attack (e.g., benzene to phenol), and the newly introduced hydroxyl function will undergo glucouronidation (e.g., phenol to phenyl glucouronic acid). Some metabolites are formed as impurities during the development of a process. Control of these process-related metabolite impurities in the final API may not be necessary if control of other metabolites has already occurred and taken into consideration. Tightening the limits, therefore, may not be needed.
The development of a new drug mandates that meaningful and reliable analytical data be generated at various steps of drug development. The drug also should exhibit excellent stability throughout its shelf-life. To meet these requirements, methodologies need to be developed that are sensitive enough to measure low levels of impurities. This need has led to analytical methods that are suitable for determining trace and ultra-trace levels (i.e., submicrogram) quantities of various chemical entities (35–39). Various methods are available for monitoring impurities.Examples are asenapine N-oxide, asenapine desmethyl, and ciprofoxacin ethyl diamino impurity, which are formed as process impurities, but are also metabolites of the same process (see Figure 5). It put forth a question whether limiting such a metabolite impurity in the final API is still required.Select analytical methodologies
Spectroscopic methods . Various spectroscopic methods can be used for characterization of impurities, such as UV-visible spectroscopy, FTIR spectroscopy, NMR spectroscopy, and mass spectrometry (MS).
Separation methods . Various separation methods can be used, including thin-layer chromatography (TLC), gas chromatography (GC), HPLC, capillary electrophoresis (CE), and supercritical fluid chromatography (SFC). A review of these methods is provided in the literature (39). CE is an electrophoretic method that is frequently lumped with chromatographic methods because it shares many of the common requirements of chromatography. A broad range of compounds can be resolved using TLC by using different plates and mobile phases. GC is a useful technique for quantification. It can provide the desired resolution, selectivity, and ease of quantification. This technique is useful for organic volatile impurities. SFC offers some of the advantages of GC in terms of detection and HPLC in terms of separation.
Hyphenated methods. The following hyphenated methods can be used effectively to monitor impurities: GC–MS; liquid chromatography (LC)–MS; LC–diode-array detection (DAD)–MS; LC–NMR; LC–DAD–NMR–MS; and LC–MS–MS.
Isolating impurities . It is often necessary to isolate impurities because the instrumental methods are not available or further confirmation is needed. The following methods have been used for isolation of impurities: solid-phase extraction, liquid–liquid extraction, accelerated solvent extraction, supercritical fluid extraction, column chromatography, flash chromatography, TLC, HPLC, CE, and SFC.

Figure 5: Process impurities, thermal decomposition impurities, and metabolites of asenapine. CAS No. is Chemical Abstracts Service number.
Impurity profiling
Ideally, an impurity profile should show all impurities in a single format to allow monitoring of any variation in the profile. The driving forces for studying an impurity profile are quality considerations and regulatory requirements.
Samples to be profiled . Impurity profiling should be done for APIs, process check of the synthesis or formulation, and final drug product.
Components in an impurity profile. Ideally, an impurity profile should show synthesis-related impurities, formulation-related impurities, degradation products, and interaction products.Crucial factors for controlling impurities in API s. Several factors are important in controlling impurities in APIs as further outlined.
Crystallization . The size of crystals not only determines the quality, but also the stability of the drug. During crystallization, fine crystals should be formed to prevent entrapment of minute amounts of chemicals from the mother liquor, which in turn causes degradation of the drug.
Wet-cake washing. Many unwanted chemicals, including residual solvents, could be removed by thorough washing of the wet cake, which if not done correctly, could lead to retention of solvents and impurities in the cake.
Drying. Use of vacuum dryer or a fluid-bed dryer is always advisable in comparison to a tray dryer. Use of the former reduces drying time and brings about uniform drying, which is helpful in drying sensitive drug substances.
Appropriate packaging. The packing of bulk drugs should be based upon their nature and sensitivity. Light-sensitive products should be packed in light protective packing. Use of opaque containers for ciprofloxacin eye-drops preparations protects the active ingredients from photodegradation (22). Use of ampuls with either black carbon paper or aluminum foil for ergometrine produced negligible degradation (40). It is important to determine the most appropriate container-closure system.
Production methods based on stability studies . A manufacturer of a bulk drug should perform a detailed investigation of the process, including stability studies while finalizing the method of preparation. For example, for producing diclofenac sodium injections, the aseptic filtration process is better than the autoclave method that produces the impurity (16).
Measures by pharmacopoeias . Pharmacopoeias should take steps to incorporate impurity limits for drug substances made from a raw material in which that particular impurity is controlled. It becomes convenient for the users if the impurity limit is mentioned in the dosage forms.
Conclusion
Parts I, II, and III of this article discussed the types, origin, causes, chemistry, and impact of impurities in APIs and drug products (1, 2). Parts I and II explained how, when, and why impurities are formed. This article, Part III, highlighted the degradation-related, formulation-related, and metabolite impurities, the various analytical techniques available for their identification and separation, and crucial factors that are to be controlled while preparing bulk drugs. |
References
1. K.R. Wadekar et al., Pharm. Technol. 36 (2), 46–51 (2012).
2. K.R. Wadekar et al., Pharm. Technol. 36 (3), 58–70 (2012).
3. ICH, Q1A (R2) Stability Testing of New Drug Substances and Products (Nov. 2003).
4. ICH, Q3A(R) Impurities in New Drug Substances (Feb. 2003).
5. ICH, Q3B (R) Impurities in New Drug Products (Nov. 2003).
6. ICH, Q3C (R5), Impurities: Guideline for Residual Solvents (March 2011).
7. K.M. Alsante et al., Am. Pharm. Rev. 4 (1), 70–78 (2001).
8. T.R. Sharp, Am. Pharm. Rev. 9 (7), 84–91 (2006).
9. T.R. Sharp, Am. Pharm. Rev. 9 (3), 100–105 (2006).
10. J.A. Mollica et al., J. Pharma. Sci. 67 (4), 443–465 (1978).
11. S. Ahuja, Impurities Evaluation of Pharmaceuticals (Marcel Dekker, New York, 1998)
12. L. Gu et al., Int. J. Pharm. 41 (1–2) 105–113 (1988).
13. P.V. Devarajan et al., J. Pharm. Biomed. Anal. 22 (4), 679–683 (2000).
14. M.C. Damle et al., J. Adv. Sci. Res. 2 (3), 77-82 (2011).
15. K.A. Connors, G.L. Amidon, and V. J. Stella, Chemical Stability of Pharmaceuticals—A Handbook for Pharmacists (John Wiley & Sons, New York, 1986).
16. J. Roy et al., J. Pharm. Sci. 90 (5) 541–544 (2001).
17. G.J.A. Walker et al., Lancet 13 (2), 393–393, (1988).
18. H.V. Hogerzeil et al., British Medical Journal 304 (25), 210–212 (1992).
19. J. Roy et al., Indian Drugs 34 (11), 634–636 (1997).
20. J. Roy et al., Drug Dev. Ind. Pharm. 20 (13), 2157–2163 (1994).
21. P.S. Rao et al., J. Pharm. Biomed. Anal. 56 (2) 413–418, (2011).
22. E. Fasani et al., Photochem. Photobiol. 68 (5) 666–674 (1998).
23. K. M Alsante et al., J. Pharm. Sci. 93 (9) 2296-2309 (2004).
24. W. Steven et al., J. Pharm. Sci. 97 (2) 883–892 (2008).
25. S.W. Baertschi et al., Pharm. Technol. 26 (2) 48–54 (2002).
26. J. Tagg et al., Biochem. Pharmacol. 16 (1) 143–153 (1967).
27. F.M. Eckenrode, J. Nat. Prod. 47 (5) 882–884 (1984).
28. J.M. Bowen et al., Anal. Chem. 53 (14) 239–2242 (1981).
29. M. Colvin, Clin. Pharmacokinetics 4 (5) 380-394 (1979).
30. Z. H. Israili et al., J. Pharmacol. Exp. Ther. 187 (1) 138–151 (1973).
31. M.A. Schwartz. et al., Drug Metab. Dispos. 1 (1) 322–331 (1973).
32. K. Kassahun et al., Drug Metab. Dispos. 1 (25) 81–93 (1996).
33. E. Stoermer et al., Drug Metab. Dispos. 10 (28) 1168–1175 (2000).
34. J. G. Slatter et al., Drug Metab. Dispos. 8 (29) 1136–1145 (2001).
35. S. Ahuja, Chromatography of Pharmaceuticals—Natural, Synthetic and Recombinant Products, ACS Symposium Series 512 (American Chemical Society, Washington, DC, 1992).
36. S. Ahuja, Trace and Ultratrace Analysis by HPLC (John Wiley & Sons, New York, 1992).
37. S. Ahuja, Chromatography and Separation Science (Elsevier, 2003).
38. S. Ahuja and S. Scypinski, Eds., Handbook of Modern Pharmaceutical Analysis(Academic Press, 2001).
39. S. Ahuja and M.Dong, Eds., Handbook of Pharmaceutical Analysis by HPLC: Volume 6 (Academic Press, 2005).
40. J. Roy et al., Indian Drugs 34 (11) 634–636 (1997).
READ
ANTHONY MELVIN CRASTO
https://newdrugapprovals.org/

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Linkedin

Join me on Facebook  FACEBOOK
FACEBOOK
Join me on twitter

 amcrasto@gmail.com
amcrasto@gmail.com
CALL +919323115463 INDIA
////////////
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
47,959 total views, 2 views today



















