











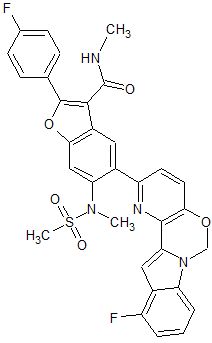

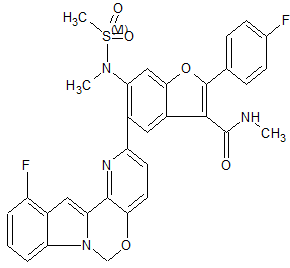
MK 8876
CAS 1426960-33-9
2-(4-Fluorophenyl)-5-(11-fluoro-6H-pyrido[2′,3′:5,6][1,3]oxazino[3,4-a]indol-2-yl)-N-methyl-6-(N-methylmethanesulfonamido)-1-benzofuran-3-carboxamide
| 2-(4-Fluorophenyl)-5-(11-fluoro-6H-pyrido[2′,3′:5,6][1,3]oxazino[3,4-a]indol-2-yl)-N-methyl-6-[methyl(methylsulfonyl)amino]-3-benzofurancarboxamide |
| Molecular Formula | C32H24F2N4O5S | |
| Molecular Weight | 614.62 |
- Originator Merck & Co
- Class Antivirals
- Phase I Hepatitis C
Most Recent Events
- 11 Oct 2013 Phase-I clinical trials in Hepatitis C in Germany (PO)
- 11 Oct 2013 Phase-I clinical trials in Hepatitis C in Moldova (PO)
- 23 Aug 2013 Preclinical trials in Hepatitis C in USA (PO)
DATA
2-(4-Fluorophenyl)-5-(11-fluoro-6H-pyrido[2′,3′:5,6][1,3]oxazino[3,4-a]indol-2-yl)-N-methyl-6-(N-methylmethanesulfonamido)-1-benzofuran-3-carboxamide
MK-8876 off-white solid
1H NMR (500 MHz, DMSO-d6) δ 8.56 (q, J = 4.7 Hz, 1H), 8.06–8.01 (m, 2H), 8.05 (s, 1H), 7.86 (s, 1H), 7.71 (d, J = 8.5 Hz, 1H), 7.62 (d, J = 8.5 Hz, 1H), 7.52 (d, J = 8.3 Hz, 1H), 7.46–7.40 (m, 2H), 7.29–7.22 (m, 1H), 7.11 (s, 1H), 6.94 (dd, J = 10.6, 7.9 Hz, 1H), 6.27 (s, 2H), 3.31 (s, 3H), 2.96 (s, 3H), 2.85 (d, J = 4.7 Hz, 3H);
13C NMR (125.7 MHz, DMSO-d6) δ 162.86, 162.82 (d, JC–F = 248.5 Hz), 155.74 (d, JC–F = 246.1 Hz), 153.80, 152.43, 152.28, 147.20, 137.08, 137.00 (d, JC–F = 10.8 Hz), 136.36, 136.20, 132.37, 129.50 (d, JC–F = 8.6 Hz), 127.17, 125.45 (d, JC–F = 3.1 Hz), 125.08, 125.02, 123.70 (d, JC–F = 7.7 Hz), 122.28, 117.23 (d, JC–F = 22.4 Hz), 116.01 (d, JC–F = 21.9 Hz), 113.65, 111.76, 106.90 (d,JC–F = 3.5 Hz), 105.32 (d, JC–F = 18.5 Hz), 94.16, 73.57, 39.39, 37.24, 26.16;
HR-ESI-MS m/zcalcd for C32H25N4O5SF2+ [M + H]+ 615.1514, found 615.1500.
| HPLC Method | |
|---|---|
| column | Ascentis Express C18 2.7 μm (fused core), 100 mm × 4.6 mm |
| detection | UV at 210 nm |
| column temperature | 40 °C |
| flow rate | 1.8 mL/min |
| injection volume | 5.0 μL |
| gradient | 90% A to 5% A over 11 min, hold at 5% A for 2 min, 5% A back to 90% A over the next 0.1 min, and then hold at 90% A for 2.9 min |
| run time | 16 min |
| data collection | acquisition for the first 13 min |
| mobile phases | solvent A: water with 0.1% H3PO4 |
| solvent B: acetonitrile | |
| Retention Time Data | |
|---|---|
| identity | tR (min) |
| boronic acid 27 | 4.24 |
| desbromoarene 28 | 5.33 |
| MK-8876 (1) | 7.89 |
| chloropyridine starting material 2 | 8.03 |
| BHT | 10.22 |
SYNTHESIS



CONTD……………
MK 8876










MK 8876
Patent
Scheme 1

Scheme 2

Scheme 3

Q
Scheme 4

EXAMPLES
Example 1
Preparation of Compound 1
 THIS COMPD HAS ONE FLUORO MISSING, APPLY TO YOUR MK 8876
THIS COMPD HAS ONE FLUORO MISSING, APPLY TO YOUR MK 8876Step 1 – Synthesis of 2,6-dichloropyridin-3-ol

Η202 (1.60 g, 47.12 mmol) was added slowly to the solution of compound 2,6- dichloropyridin-3-ylboronic acid (3 g, 15.71 mmol) in CH2CI2 (30 mL) at 0 °C. After stirred at room temperature for about 15 hours, the mixture was quenched with sat. Na2S203 aqueous (50 mL) and adjusted to pH < 7 with IN HC1. The mixture was extracted with EtOAc (40 mL x 3). The organic layer was washed with brine (100 mL), dried over Na2S04, filtered and the solvent was evaporated to provide2,6-dichloropyridin-3-ol (2.34 g, yield: 91.4%). 1H-NMR (CDC13, 400 MHz) δ 7.30 (d, / = 8.4 Hz, 1H), 7.19 (d, / = 8.4 Hz, 1H), 5.70 (br, 1H).
– Synthesis of 2,6-dichloro- -methoxypyridine
To a solution of 2,6-dichloropyridin-3-ol (16.3 g, 0.1 mol) and K2C03 (41.4 g, 0.3 mol) in DMF (200 mL) were added Mel (21.3 g, 0.15 mol). The mixture was allowed to stir at 80 °C for 2 hours. The mixture was then diluted with water (200 mL) and extracted with EtOAc (200 mL x 3). The organic layer was washed with brine (200 mL x 3), dried over Na2S04, filtered and the solvent was evaporated to provide 2,6-dichloro-3-methoxypyridine (17.0 g, yield: 96.0%). 1H-NMR (CDC13, 400 MHz) δ 7.12-7.18 (m, 2H), 3.86 (s, 3H). Step 3 – Synthesis of2-(6-chloro-3-methoxypyridin-2-yl)-lH-indole

To a degassed solution of compound 2,6-dichloro-3-methoxypyridine (8.9 g, 0.05 mol), (l-(tert-butoxycarbonyl)-lH-indol-2-yl)boronic acid (13 g, 0.05 mol) and K3PO4 (31.8 g, 3.0 mol) in DMF (100 mL) was added Pd(dppf)Cl2 (3.65 g, 0.005 mol) under N2. The mixture was heated at 60 °C for about 15 hours. The reaction mixture was cooled to room temperature, diluted with EtOAc and filtered. The filtrate was washed with H20, brine, dried over Na2S04. After being concentrated in vacuo, the resulting residue was purified using prep-HPLC to provide the desired product of 2-(6-chloro-3-methoxypyridin-2-yl)-lH-indole (9.0 g, yield:
69.8%). 1H-NMR (CDC13, 400 MHz) δ 9.52 (s, 1H), 7.65 (d, / = 7.6 Hz, 1H), 7.38-7.43 (m, 2H), 7.07-7.26 (m, 4H), 4.03 (s, 3H).
Step 4 – Synthesis of6-chlor -2-(lH-indol-2-yl)pyridin-3-ol

BBr3 (0.4 mL, 0.39 mmol) was added to the solution of 2-(6-chloro-3- methoxypyridin-2-yl)-lH-indole (50 mg, 0.194 mmol) in CH2C12 (0.5 mL) at -78 °C under N2. The mixture was allowed to stir at room temperature for 3 hours. The mixture was then quenched with CH3OH (10 mL) at -78 °C. After being concentrated in vacuo, the resulting residue was purified using prep-TLC (PE : EtOAc = 2.5 : 1) to afford the desired product of 6- chloro-2-(lH-indol-2-yl)pyridin-3-ol (40 mg, yield: 85.1%). 1H-NMR (CDC13, 400 MHz) δ 10.09 (s, 1H), 9.72 (s, 1H), 7.50 (d, / = 7.9 Hz, 1H), 7.17-7.32 (m, 3H), 7.08-7.14 (m, 1H), 6.87-6.96 (m, 2H).
Step 5 – Synthesis of 2-chlo -6H-pyrido[2′ ,3′ : 5 ,6] [ 1 ,3]oxazino[3 ,4-a]indole

To a solution of chloroiodomethane (3.51 g, 20.0 mmol) and K2CO3 (1.38 g, 10.0 mmol) in DMF (50 mL) was allowed to stir at 100 °C, 6-chloro-2-(lH-indol-2-yl)pyridin-3-ol (480 mg, 2.0 mmol) in DMF (50 mL) was added dropwise. After addition, the mixture was allowed to stir for another 0.5 hours. The mixture was then diluted with water (100 mL) and extracted with EtOAc (100 mL x 3). The organic layer was washed with brine (100 mL x 3), dried over Na2S04 and concentrated. The residue was purified using prep-TLC (PE : EtOAc = 3 1) to afford the desired product of 2-chloro-6H-pyrido[2′,3′:5,6][l,3]oxazino[3,4-a]indole (260 mg, yield: 50.7%). 1H-NMR (CDC13, 400 MHz) δ 7.63 (d, / = 8.0 Hz, 1H), 7.22-7.27 (m, 3H), 7.19 (d, / = 2.4 Hz, 1H), 7.08-7.12 (m, 2H), 5.86 (s, 2H).
Step 6 – Synthesis of2-(4-fluowphenyl)-N-methyl-6-(N-methylmethylsulfonamido)-5-(6H- pyridol 2 ‘,3’:5,6][ l, mpound 1 )
 THIS COMPD HAS ONE FLUORO MISSING, APPLY TO YOUR MK 8876
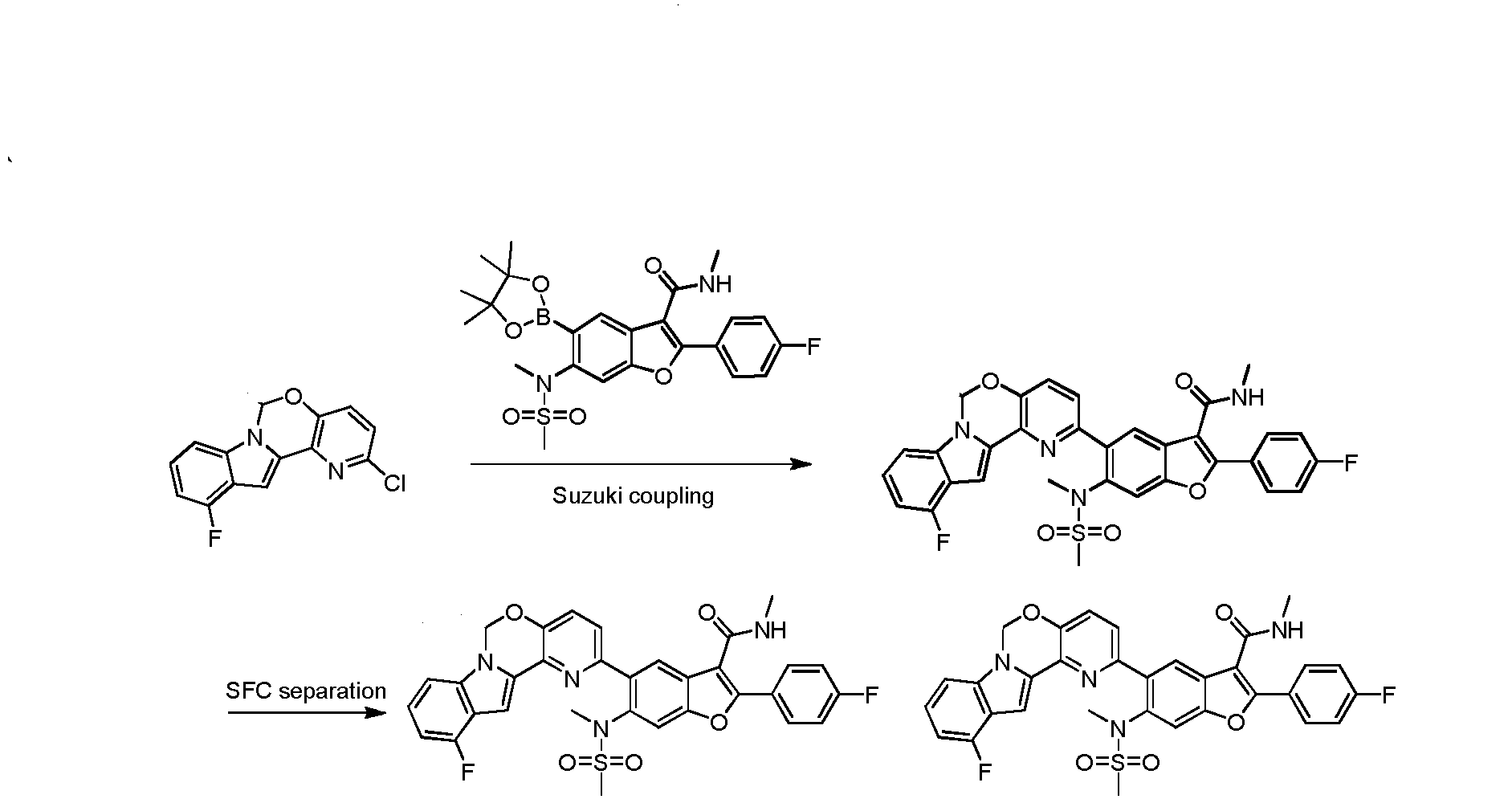
THIS COMPD HAS ONE FLUORO MISSING, APPLY TO YOUR MK 8876To a degassed solution of 2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzofuran-3- carboxamide (502 mg, 1.0 mmol), 2-chloro-6H-pyrido[2′,3′:5,6][l,3]oxazino[3,4-a]indole (256 mg, 1.0 mmol) and K3PO4 (636 mg, 3.0 mmol) in dioxane : H20 (1.5 mL : 0.4 mL) was added Pd2(dba)3 (91 mg, 0.1 mmol) and X-phos (91 mg, 0.2 mmol) under N2. The mixture was heated to 110 °C for 3 hours. The reaction mixture was cooled to room temperature, diluted with EtOAc and filtered. The filtrate was washed with H20, brine, dried over Na2S04. After being concentrated in vacuo, the resulting residue was purified using prep-HPLC to provide the desired product of Compound 1 (275 mg, yield: 46.1%). 1H-NMR (CDC13, 400 MHz) δ 7.88-7.94 (m, 3H), 7.61-7.63 (m, 2H), 7.40 (s, 2H), 7.09-7.28 (m, 6H), 5.94 (s, 2H), 5.86 (d, / = 4.4 Hz, 1H), 3.29 (s, 3H), 2.92 (d, / = 5.2 Hz, 3H), 2.65 (s, 3H). MS (M+H)+: 596.
Compounds 2-15, depicted in the table below, were prepared using the method described above.
COMPD 2 IS MK 8876

PATENT
Example 81
Preparation of Compound 2
Synthesis of ethyl 3- 4-fluorophenyl)-3-oxopropanoate
Diethyl carbonate (130 g, 1.1 mol) was dissolved in a suspension ofNaH (60% in oil, 50.2 g, 1.3 mol) in anhydrous tetrahydrofuran (1.5 L), and then l-(4-fluorophenyl)ethanone (150 g, 1.09 mol) was added dropwise at 70 °C. The resulting mixture was stirred at 70 °C for 3 hours. After the reaction mixture was cooled to room temperature and poured into HCl (1 N). The mixture was extracted with EtOAc, the organic phase was dried with anhydrous NaS04 and concentrated in vacuo. The resulting residue was purified using column chromatography (eluted with petroleum ether / EtOAc = 50 / 1) to provide ethyl 3-(4-fluorophenyl)-3-oxopropanoate (217 g, yield: 95%). 1H-NMR (CDC13, 400 MHz) δ 7.92-7.97 (m, 2H), 7.07-7.13 (m, 2H), 4.14-4.20 (m, 2H), 3.93 (s, 2H), 1.22 (d, J= 7.2 Hz, 3H). MS (M+H)+: 211. Step 2 – Synthesis of ethyl 5-bromo-2-(4-fluorophenyl)benzofuran-3-carboxylate
A solution of ethyl 3-(4-fluorophenyl)-3-oxopropanoate (130 g, 0.6 mol), 4- bromophenol (311 g, 1.8 mol) and FeCl3-6H20 (19.5 g, 0.09 mol) in DCE (700 mL) was heated to reflux, and then 2-(tert-butylperoxy)-2-methylpropane (193 g, 1.32 mol) was added dropwise under nitrogen. After 6 hours of refluxing, the mixture was cooled to RT, quenched with saturated NaHS03 and extracted with dichloromethane. The organic phases were washed with water, brine and dried over Na2S04, filtered and concentrated in vacuo. The resulting residue was purified using column chromatography (petroleum ether / dichloromethane = 15 / 1) to provide the crude product, which was crystallized from cold MeOH to provde ethyl 5-bromo-2- (4-fluorophenyl)benzofuran-3-carboxylate (37 g, yield: 14.3%) as solid. 1H- MR (CDC13, 400 MHz) δ 8.12 (s, 1H), 7.97-8.01 (m, 2H), 7.37 (d, J= 4.0 Hz, 1H), 7.32 (d, J= 8.0 Hz, 1H), 7.11 (t, J= 8.0 Hz, 2H), 4.32-4.38 (m, 2H), 1.36 (t, J= 8.0 Hz, 3H). MS (M+H)+: 363 / 365.
Step 3 – Synthesis of eth l 5-bromo-2-(4-fluorophen -6-nitrobenzofuran-3-carboxylate
To a solution of ethyl 5-bromo-2-(4-fluorophenyl)benzofuran-3-carboxylate (50 g,
137.6 mmol) in CHC13 (500 mL), fuming HN03 (50 mL) was added dropwise at -15 °C and the mixture was stirred for 0.5 hour. The reaction mixture was poured into ice water and extracted with CH2C12. The organic layer was washed with a.q. sat. NaHC03 and brine, after removed the most of solvent, the resulting residue was crystallized with petroleum ether / dichloromethane = 20 / 1 to provide product of ethyl 5-bromo-2-(4-fluorophenyl)-6-nitrobenzofuran-3-carboxylate (35 g, yield: 66%). 1H- MR (CDC13, 400 MHz) δ 8.36 (s, 1H), 8.02-8.04 (m, 3H), 7.13-7.18 (m, 2H), 4.36-4.41 (m, 2H), 1.37 (t, J= 4.0 Hz, 3H). MS (M+H)+: 408 / 410.
Step 4 – Synthesis of ethyl 6-amino-5-bromo-2-(4-fluorophenyl)benzofuran-3-carboxylate
A mixture of ethyl 5-bromo-2-(4-fluorophenyl)-6-nitrobenzofuran-3-carboxylate (52 g, 127 mmol), iron filings (21.3 g, 382.2 mmol) and H4C1 (41 g, 764.4 mmol) in MeOH / THF / H20 (2 / 2 / 1, 500 mL) was stirred at reflux for 3 hour. After filtered and concentrated, the resulting residue was purified using column chromatography (petroleum ether / EtOAc / dichloromethane = 20 : 1 : 20) to provide ethyl 6-amino-5-bromo-2-(4-fluorophenyl) benzofuran-3-carboxylate (40 g, yield: 82%). 1H- MR (CDC13, 400 MHz) δ 8.01 (s, 1H), 7.94-7.98 (m, 2H), 7.08 (t, J= 8.0 Hz, 2H), 6.83 (s, 1H), 4.32-4.36 (m, 2H), 4.18 (s, 2H), 1.35 (t, J= 8.0 Hz, 3H). MS (M+H)+: 378 / 380.
Step 5 – Synthesis of 5-Bromo-2-(4-fluoro-phenyl)-6-methanesulfonylamino-benzofuran-3- carboxylic acid eth l ester
MsCI (31.7 g, 277.5 mmol) was added to a solution of ethyl 6-amino-5-bromo-2- (4-fluorophenyl)benzofuran-3-carboxylate (35 g, 92.5 mmol) and pyridine (60 mL) in
dichloromethane (300 mL) at 0 °C. After stirred overnight at room temperature, the mixture was diluted with water and extracted with dichloromethane. The organic layer was washed with brine, dried over Na2S04, filtered and concentrated in vacuo, the resulting residue was purified using crystallized with EtOAc to provde the pure product of ethyl 5-bromo-2-(4-fluorophenyl)-6- (methylsulfonamido)benzofuran-3-carboxylate (35 g, yield: 82%). 1H- MR (CDC13, 400 MHz) δ 8.27 (s, 1H), 8.01-8.05 (m, 2H), 7.87 (s, 1H), 7.15-7.19 (m, 2H), 6.87 (s, 1H), 4.38-4.43 (m, 2H), 3.00 (s, 3H), 1.40 (t, J= 40 Hz, 3H). MS (M+H)+: 456 / 458.
Step 6 – Synthesis of 5-Bromo-2-(4-fluoro-phenyl)-6-methanesulfonylamino-benzofuran-3- carboxylic acid
To a solution of ethyl 5-bromo-2-(4-fluorophenyl)-6-(methylsulfonamido) benzofuran-3-carboxylate (53 g, 0.23 mol) in dioxane / H20 (5 / 1, 600 mL) was added
LiOH-H20 (25 g, 1.17 mol), and the mixture was stirred at 100 °C for 3 hours. After
concentrated, the resulting residue was dissolved in H20, 1 N HCl was added until pH reached 3, and the mixture was extracted with EtOAc. The organic layer was washed with brine, dried over Na2S04 and filtered. The solvent was removed to provide the product of 5-bromo-2-(4- fluorophenyl)-6-(methylsulfonamido)benzofuran-3-carboxylic acid (48 g, yield: 96%).1H- MR (DMSO- e, 400 MHz) δ 13.49 (s, 1H), 9.67 (s, 1H), 8.30 (s, 1H), 8.12-8.17 (m, 2H), 7.87 (s, 1H), 7.45-7.50 (m, 2H), 3.16 (s, 3H). MS (M+H)+: 428 / 430. Step 7 – Synthesis of 5-Bromo-2-(4-fluoro-phenyl)-6-methanesulfonylamino-benzofuran-3- carboxylic acid methylamide

A solution of 5-bromo-2-(4-fluorophenyl)-6-(methylsulfonamido) benzofuran-3- carboxylic acid (33 g, 77 mmol), HOBT (15.6 g, 115.5 mmol) and EDCI (22.2 g, 115.5 mmol) in DMF (250 mL) was stirred at room temperature. After 2 hours, Et3N (50 mL) and CH3 H2 (HC1 salt, 17.7 g, 231 mmol) was added to the mixture, and the mixture was stirred overnight. After the solvent was removed, H20 was added and the mixture was extracted with ethyl acetate. The combined organic layer was washed with H20, brine and concentrated in vacuo. The resulting residue was washed with EtOAc to provide the product of 5-bromo-2-(4-fluorophenyl)-N- methyl-6-(methylsulfonamido)benzofuran-3-carboxamide (32 g, yield: 94%). 1H- MR (DMSO- ck, 400 MHz) δ 9.55 (br s, 1H), 8.46-8.48 (m, 1H), 8.12-8.17 (m, 2H), 7.96 (s, 1H), 7.87 (s, 1H), 7.45-7.50 (m, 2H), 3.16 (s, 3H), 2.93 (d, J= 8.4 Hz, 3H). MS (M+H)+: 441 / 443.
Step 8 – Synthesis of 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido benzofuran-3-carboxamide

CH3I (31.6 g, 223 mmol) was added to a mixture of 5-bromo-2-(4-fluorophenyl)- N-methyl-6-(methylsulfonamido)benzofuran-3-carboxamide (32 g, 74 mmol), K2C03 (25.6 g, 186 mmol) and KI (246 mg, 1.5 mmol) in DMF (150 mL) under N2 protection. The mixture was stirred at 80-90 °C overnight. After concentrated in vacuo, the resulting residue was washed with water (200 mL) and EtOAc (200 mL) to provide the product of 5-bromo-2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide (31.5 g, 94%). 1H- MR (CDCI3, 400 MHz) δ 8.16 (s, 1H), 7.88-7.92 (m, 2H), 7.70 (s, 1H), 7.18-7.23 (m, 2H), 5.78 (br s, 1H), 3.34 (s, 3H), 3.09 (s, 3H), 3.00 (d, J= 4.8 Hz, 3H). MS (M+H)+: 455 / 457. Step 9 – Synthesis of 2-(4-fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)-5-(4, 4, 5, 5- tetramethyl-1 -dioxaborolan-2-yl)benzofuran-3-carboxamide
a degassed solution of 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide (1.0 g, 2.2 mmol) and pinacol diborane (2.79 g, 11.0 mmol) in 1,4-Dioxane (25 mL) was added KOAc (647 mg, 6.6 mmol) under N2 and stirred for 4 hours at room temperature. Then Pd(dppf)Cl2 (60 mg) was added, and the mixture was stirred for another 30 minutes. Then the mixture was put into a pre-heated oil-bath at 130 °C and stirred for another 1 hour under N2. The reaction mixture was cooled to room
temperatureand concentrated and extracted with EtOAc. The organic layers were washed with brine, dried over Na2S04. After concentrated, the crude product of the boronic ester was purified using column chromatography (petroleum ether / EtOAc = 5 / 1 to 2 / 1) to obtain 2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)-5-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)benzofuran-3-carboxamide as white solid (700 mg, yield: 64%). 1H- MR (CDCI3, 400 ΜΗζ) δ 8.17 (s, 1H), 7.87-7.91 (m, 2H), 7.52 (s, 1H), 7.11 (t, 7= 7.6 Hz, 2H), 5.81 (d, 7= 2.8 Hz, 1H), 3.30 (s, 3H), 2.97 (d, 7= 5.2 Hz, 3H), 2.90 (s, 3H), 1.31 (s, 12H). MS (M+H)+: 503.
Step 10 – Synthesis of tert-butyl 4-fluoro-lH-indole-l -car boxy late
To a solution of 4-fluoro-lH-indole (5 g, 0.11 mol) and DMAP (150 mg, 3%Wt) in THF (50 mL) was added (Boc)20 (8.5 g, 0.04 mol) dropwise. The mixture was stirred at room temperature for 2 hours. The organic solvent was removed in vacuo, and the resulting residue was purified using column chromatography (pure petroleum ether) to provide tert-butyl 4-fluoro- lH-indole-l-carboxylate (8.3 g, yield: 96%). 1H- MR (CDC13, 400 MHz) δ 7.92 (d, J= 8.4 Hz, 1H), 7.55 (d, J= 3.6 Hz, 1H), 7.23 (m, 1H), 6.90 (m, 1H), 6.66 (d, J= 3.6 Hz, 1H), 1.67 (s, 9H). MS (M+H)+: 236.
Step 11 – Synthesis of (l-(tert-butoxycarbonyl)-4-fluoro-lH-indol-2-yl)boronic acid

To a solution of diisopropylamine (7.5 mL, 0.11 mol) in THF (35 mL) at 0 °C was added «-BuLi (21 mL, 0.055 mol) dropwise. The mixture was stirred at 0 °C for 40 minutes. Then the mixture was cooled to -78 °C. Tert-butyl 4-fluoro-lH-indole-l-carboxylate (5 g, 0.02 mol) in THF (13 mL) was added dropwise slowly. After addition, the mixture was stirred at -78 °C for 2 hours. Then triisopropyl borate (3.29 g, 0.03 mol) was added. The mixture was stirred at -78 °C for another 40 minutes. The reaction was monitored using TLC. When the reaction was completed, the mixture was adjusted to pH = 6 with 1 N HC1. After extracted with EtOAc (25 mL x 3), the combined organic layers were washed with brine (50 mL), dried over Na2S04, filtered and concentrated in vacuo. The obtained solid was recrystallized with EtOAc and petroleum ether to provide (l-(tert-butoxycarbonyl)-4-fluoro-lH-indol-2-yl)boronic acid (4.5 g, yield: 76.7%, which might be unstable at high temp, work up, store in fridge). 1H- MR (CDC13, 400 MHz) δ 7.77 (d, J= 8.4 Hz, 1H), 7.57 (s, 1H), 7.44 (s, 2H), 7.24 (m, 1H), 6.90 (m, 1H), 1.66 (s, 9H). MS (M+H)+: 280.
Step 12 – Synthesis of 6-chloro-2-iodopyridin-3-ol
6-chloropyridin-3-ol (5.0 g, 38.6 mmol) was dissolved in water (50 mL) and placed under an N2 atmosphere. Na2C03 (8.2 g, 77.4 mmol) was added followed by iodine (9.8 g, 38.8 mmol). The reaction mixture was stirred at room temperature for 2 hours. The mixture was poured into 1M Na2S203 and extracted with EtOAc. The combined organic phases were washed with brine, dried over Na2S04 and concentrated to provide the product of 6-chloro-2- iodopyridin-3-ol (7.0 g, yield: 70.9%). 1H- MR (CDC13, 400 MHz) δ 7.17 (d, J= 8.4 Hz, 1H), 7.06 (d, J= 8.4 Hz, 1H). MS (M+H)+: 256 / 258.
Step 13 – Synthesis of 6-chloro-2-(4-fluoro-lH-indol-2-yl)pyridin-3-ol
A mixture of (l-(tert-butoxycarbonyl)-4-fluoro-lH-indol-2-yl)boronic acid (5 g, 18.0 mmol), 6-chloro-2-iodopyridin-3-ol (3.82 g, 15.0 mol) and NaHC03 (3.78 g, 45.0 mol) in 1, 4-dioxane (76 mL) and water (7 mL) was stirred at room temperature for 15 minutes. Then Pd(PPh3)2Cl2 (527 mg, 0.75 mmol) was added under nitrogen atmosphere, and the mixture was heated at 100 °C under N2 for 16 hours. The reaction mixture was cooled to room temperature, diluted with EtOAc (50 mL), filtered and concentrated in vacuo. The resulting residue was diluted with H20 (60 mL) and EtOAc (30 mL), and the layer was separated, the aqueous layer was extracted with EtOAc (3*30 mL). The combined organic layers were washed with brine (50 mL), dried over Na2S04, filtered and concentrated in vacuo. The resulting residue was purified using column chromatography (petroleum ether / EtOAc = 20 / 1 ~ 3 / 1) to provide 6-chloro-2- (4-fluoro-lH-indol-2-yl)pyridin-3-ol (3 g, yield: 76.5%). 1H- MR (MeOD, 400 MHz) δ 7.36 (s, 1H), 7.23-7.27 (m, 2H), 7.03-7.11 (m, 2H), 6.63-6.68 (m, 1H). MS (M+H)+: 263 / 265.
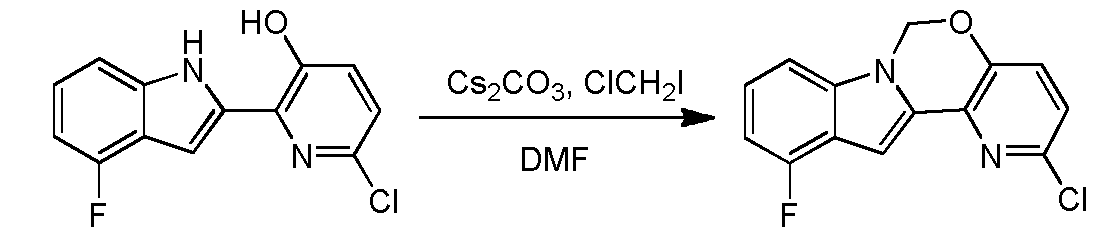
Ste 14 – Synthesis of 2-chloro-ll-fluoro-6H-pyrido[2′,3′:5, 6][l,3]oxazino[3,4-a]indole
A solution of 6-chloro-2-(4-fluoro-lH-indol-2-yl)pyridin-3-ol (2 g, 7.6 mmol) and Cs2C03 (7.46 g, 22.89 mmol) in DMF (100 mL) was stirred at 100 °C (internal temperature) for 15 min, and then chloroiodomethane (2.85 g, 15.3 mmol) in DMF (2 mL) was added dropwise. After the reaction was completed, the mixture was filtered and concentrated in vacuo. The resulting residue was diluted with water (50 mL) and extracted with ethyl acetate (30 mL x 3). The organic layer was washed with brine, dried over Na2S04 and concentrated in vacuo. The resulting residue was purified using column chromatography (petroleum ether:EA=10: l) to provde 2-chloro-l l-fluoro-6H-pyrido[2′,3′:5,6][l,3]oxazino[3,4-a]indole (1.8 g, yield: 86.1%). 1H- MR (DMSO-i¾, 400 MHz) δ 7.64 (d, J= 8.8 Hz, 1H), 7.39-7.46 (m, 2H), 7.21-7.25 (m, 1H), 7.06 (s, 1H), 6.88-6.92 (m, 1H), 6.18 (s, 2H). MS (M+H)+: 275 / 277. Step 15 – Synthesis of5-(ll-fluoro-6H-pyrido[2 3′:5, 6][l,3]oxazino[3,4-a]indol-2-yl)-2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxam
To a degassed solution of 2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzofuran-3- carboxamide (100 mg, 0.199 mmol), 2-chloro-l l-fluoro-6H-pyrido[2′,3′:5,6][l,3]oxazino[3,4- a]indole (56 mg, 0.199 mmol) and Κ3Ρ04·3Η20 (159 mg, 0.597 mmol) in dioxane / H20 (0.8 mL / 0.2 mL) was added Pd2(dba)3 (9 mg, 0.01 mmol) and X-Phos (9 mg, 0.02 mmol) under N2. The mixture was heated at 80 °C for 1 hour. The mixture was then diluted with water (30 mL) and extracted with EtOAc (15 mL x 3). The organic layer was washed with brine (20 mL), dried over Na2S04 and concentrated in vacuo. The resulting residue was purified using prep-TLC (petroleum ether / EtOAc = 1 : 1.5) to provde the pure product of 5-(l l-fluoro-6H- pyrido [2′, 3 ‘ : 5 , 6] [ 1 , 3 ]oxazino [3 ,4-a]indol-2-yl)-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide (60 mg, 48.8%). 1H- MR (CDC13, 400 MHz) δ: 7.99 (s, 1H), 7.93-7.96 (m, 2H), 7.65 (s, 1H), 7.45-7.50 (m, 2H), 7.17-7.21 (m, 4H), 7.10 (d, J= 8.0 Hz, 1H), 6.81-6.85 (m, 1H), 5.98 (s, 3H), 3.35 (s, 3H), 2.98 (d, J= 4.8 Hz, 3H), 2.72 (s, 3H). MS (M+H)+: 615.
Paper

We describe the route development and multikilogram-scale synthesis of an HCV NS5B site D inhibitor, MK-8876. The key topics covered are (1) process improvement of the two main fragments; (2) optimization of the initially troublesome penultimate step, a key bis(boronic acid) (BBA)-based borylation; (3) process development of the final Suzuki–Miyaura coupling; and (4) control of the drug substance form. These efforts culminated in a 28 kg delivery of the desired active pharmaceutical ingredient.
Process Development of the HCV NS5B Site D Inhibitor MK-8876
*E-mail: qinghao.chen@merck.com
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00405
PAPER

Using the Teasdale method, purge factor estimates for six impurities identified as mutagenic alerts in the synthesis of MK-8876 are compared to actual measured amounts of these impurities determined via appropriate analytical methods. The results from this comparison illustrate the conservative nature of purge factor estimates, meaning that overprediction of mutagenic impurity purging is unlikely when using this method. Industry and regulatory acceptance of the purge factor estimation method may help minimize analytical burden in pharmaceutical development projects.
Evaluation and Control of Mutagenic Impurities in a Development Compound: Purge Factor Estimates vs Measured Amounts
*E-mail: mark_mclaughlin@merck.com.
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00263?journalCode=oprdfk
| WO2004041201A2 * | Oct 31, 2003 | May 21, 2004 | Viropharma Incorporated | Benzofuran compounds, compositions and methods for treatment and prophylaxis of hepatitis c viral infections and associated diseases |
| WO2011106992A1 * | Mar 2, 2011 | Sep 9, 2011 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis c virus ns5b polymerase |
| WO2004041201A2 * | Oct 31, 2003 | May 21, 2004 | Viropharma Incorporated | Benzofuran compounds, compositions and methods for treatment and prophylaxis of hepatitis c viral infections and associated diseases |
| WO2010030592A1 * | Sep 8, 2009 | Mar 18, 2010 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| WO2011106992A1 * | Mar 2, 2011 | Sep 9, 2011 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis c virus ns5b polymerase |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014123794A1 * | Feb 3, 2014 | Aug 14, 2014 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds and methods of use thereof for the treatment of hepatitis c |
| WO2014123795A2 * | Feb 3, 2014 | Aug 14, 2014 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds and methods of use thereof for the treatment of hepatitis c |
| WO2014123795A3 * | Feb 3, 2014 | Oct 30, 2014 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds and methods of use thereof for the treatment of hepatitis c |
| US9242998 | Feb 3, 2014 | Jan 26, 2016 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds and methods of use thereof for the treatment of hepatitis C |
//////MK-8876, 1426960-33-9, Merck & Co, Antivirals, Phase I, Hepatitis C
Fc7cccc6c7cc2n6COc1ccc(nc12)c3cc4c(cc3N(C)S(C)(=O)=O)oc(c4C(=O)NC)c5ccc(F)cc5

Sorry, the comment form is closed at this time.