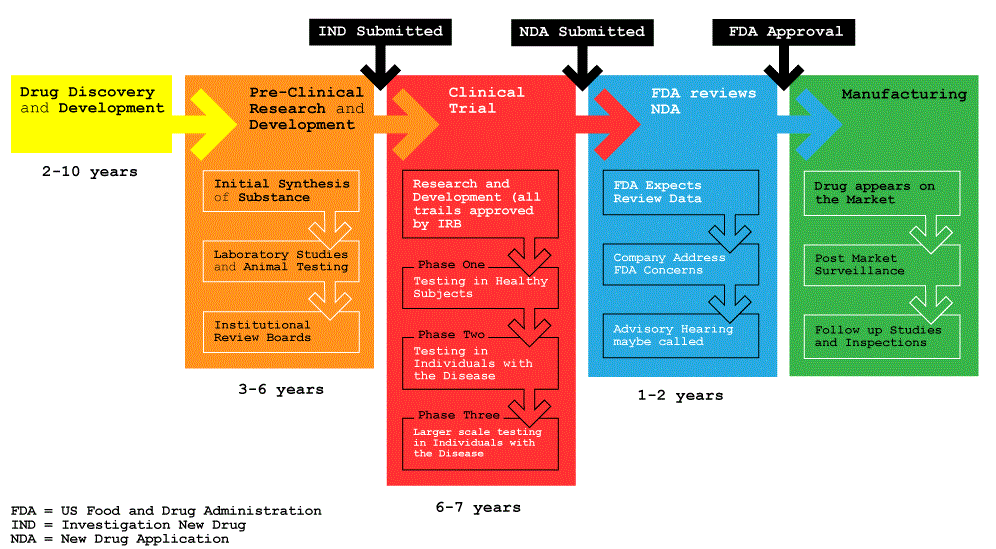
FDA’s Drug Review Process













How Drugs are Developed and Approved
The mission of FDA’s Center for Drug Evaluation and Research (CDER) is to ensure that drugs marketed in this country are safe and effective. CDER does not test drugs, although the Center’s Office of Testing and Research does conduct limited research in the areas of drug quality, safety, and effectiveness.
CDER is the largest of FDA’s five centers. It has responsibility for both prescription and nonprescription or over-the-counter (OTC) drugs. For more information on CDER activities, including performance of drug reviews, post-marketing risk assessment, and other highlights, please see the CDER Update: Improving Public Health Through Human Drugs The other four FDA centers have responsibility for medical and radiological devices, food, and cosmetics, biologics, and veterinary drugs.
Some companies submit a new drug application (NDA) to introduce a new drug product into the U.S. Market. It is the responsibility of the company seeking to market a drug to test it and submit evidence that it is safe and effective. A team of CDER physicians, statisticians, chemists, pharmacologists, and other scientists reviews the sponsor’s NDA containing the data and proposed labeling.

The section below entitled From Fish to Pharmacies: The Story of a Drug’s Development, illustrates how a drug sponsor can work with FDA’s regulations and guidance information to bring a new drug to market under the NDA process.

From Fish to Pharmacies: A Story of Drug Development
Osteoporosis, a crippling disease marked by a wasting away of bone mass, affects as many as 2 million American, 80 percent of them women, at an expense of $13.8 billion a year, according to the National Osteoporosis Foundation., The disease may be responsible for 5 million fractures of the hip, wrist and spine in people over 50, the foundation says, and may cause 50,000 deaths. Given the pervasiveness of osteoporosis and its cost to society, experts say it is crucial to have therapy alternatives if, for example, a patient can’t tolerate estrogen, the first-line treatment.
Enter the salmon, which, like humans, produces a hormone called calcitonin that helps regulate calcium and decreases bone loss. For osteoporosis patients, taking salmon calcitonin, which is 30 times more potent than that secreted by the human thyroid gland, inhibits the activity of specialized bone cells called osteoclasts that absorb bone tissue. This enables bone to retain more bone mass.
Though the calcitonin in drugs is based chemically on salmon calcitonin, it is now made synthetically in the lab in a form that copies the molecular structure of the fish gland extract. Synthetic calcitonin offers a simpler, more economical way to create large quantities of the product.
FDA approved the first drug based on salmon calcitonin in an injectable. Since then, two more drugs, one injectable and one administered through a nasal spray were approved. An oral version of salmon calcitonin is in clinical trials now. Salmon calcitonin is approved only for postmenopausal women who cannot tolerate estrogen, or for whom estrogen is not an option.
How did the developers of injectable salmon calcitonin journey “from fish to pharmacies?”
After obtaining promising data from laboratory studies, the salmon calcitonin drug developers took the next step and submitted an Investigational New Drug (IND) application to CDER. The IND Web page explains the need for this application, the kind of information the application should include, and the Federal regulations to follow.
Once the IND application is in effect, the drug sponsor of salmon calcitonin could begin their clinical trials. After a sponsor submits an IND application, it must wait 30 days before starting a clinical trial to allow FDA time to review the prospective study. If FDA finds a problem, it can order a “clinical hold” to delay an investigation, or interrupt a clinical trial if problems occur during the study.
Clinical trials are experiments that use human subjects to see whether a drug is effective, and what side effects it may cause. The Running Clinical Trials Webpage provides links to the regulations and guidelines that the clinical investigators of salmon calcitonin must have used to conduct a successful study, and to protect their human subjects.
The salmon calcitonin drug sponsor analyzed the clinical trials data and concluded that enough evidence existed on the drug’s safety and effectiveness to meet FDA’s requirements for marketing approval. The sponsor submitted a New Drug Application (NDA) with full information on manufacturing specifications, stability and bioavailablility data, method of analysis of each of the dosage forms the sponsor intends to market, packaging and labeling for both physician and consumer, and the results of any additional toxicological studies not already submitted in the Investigational New Drug application. The NDA Web page provides resources and guidance on preparing the NDA application, and what to expect during the review process.

New drugs, like other new products, are frequently under patent protection during development. The patent protects the salmon calcitonin sponsor’s investment in the drug’s development by giving them the sole right to sell the drug while the patent is in effect. When the patents or other periods of exclusivity on brand-name drugs expire, manufacturers can apply to the FDA to sell generic versions. TheAbbreviated New Drug Applications (ANDA) for Generic Drug Products Webpageprovides links to guidances, laws, regulations, policies and procedures, plus other resources to assist in preparing and submitting applications.
Bringing Nonprescription Drug Products to the Market Under an OTC Monograph
OTC drugs can be brought to the market following the NDA process as described above or under an OTC monograph. Each OTC drug monograph is a kind of “recipe book” covering acceptable ingredients, doses, formulations, labeling, and, in some cases, testing parameters. OTC drug monographs are continually updated to add additional ingredients and labeling as needed. Products conforming to a monograph may be marketed without FDA pre-approval. The NDA and monograph processes can be used to introduce new ingredients into the OTC marketplace. For example, OTC drug products previously available only by prescription are first approved through the NDA process and their “switch” to OTC status is approved via the NDA process. OTC ingredients marketed overseas can be introduced into the U.S. market via a monograph under a Time and Extent Application (TEA) as described in 21 CFR 330.14. For a more thorough discussion of how OTC drug products are regulated visit FDA laws, regulations and guidances that affect small business. Information is also provided on financial assistance and incentives that are available for drug development.

CDER Small Business and Industry Assistance (CDER SBIA)
Drug sponsors which qualify as small businesses can take advantage of special offices and programs designed to help meet their unique needs. The CDER Small Business and Industry Assistance (CDER SBIA) Webpage provides links to FDA laws, regulations and guidances that affect small business. Information is also provided on financial assistance and incentives that are available for drug development.
| |
The path a drug travels from a lab to your medicine cabinet is usually long, and every drug takes a unique route. Often, a drug is developed to treat a specific disease. An important use of a drug may also be discovered by accident.
For example, Retrovir (zidovudine, also known as AZT) was first studied as an anti-cancer drug in the 1960s with disappointing results. Twenty years later, researchers discovered the drug could treat AIDS, and Food and Drug Administration approved the drug, manufactured by GlaxoSmithKline, for that purpose in 1987.
Most drugs that undergo preclinical (animal) testing never even make it to human testing and review by the FDA. The drugs that do must undergo the agency’s rigorous evaluation process, which scrutinizes everything about the drug–from the design of clinical trials to the severity of side effects to the conditions under which the drug is manufactured.



Investigational New Drug Application (IND)–The pharmaceutical industry sometimes seeks advice from the FDA prior to submission of an IND.
Sponsors–companies, research institutions, and other organizations that take responsibility for developing a drug. They must show the FDA results of preclinical testing in laboratory animals and what they propose to do for human testing. At this stage, the FDA decides whether it is reasonably safe for the company to move forward with testing the drug in humans.

Clinical Trials–Drug studies in humans can begin only after an IND is reviewed by the FDA and a local institutional review board (IRB). The board is a panel of scientists and non-scientists in hospitals and research institutions that oversees clinical research.
IRBs approve the clinical trial protocols, which describe the type of people who may participate in the clinical trial, the schedule of tests and procedures, the medications and dosages to be studied, the length of the study, the study’s objectives, and other details. IRBs make sure the study is acceptable, that participants have given consent and are fully informed of their risks, and that researchers take appropriate steps to protect patients from harm.

Phase 1 studies are usually conducted in healthy volunteers. The goal here is to determine what the drug’s most frequent side effects are and, often, how the drug is metabolized and excreted. The number of subjects typically ranges from 20 to 80.

Phase 2 studies begin if Phase 1 studies don’t reveal unacceptable toxicity. While the emphasis in Phase 1 is on safety, the emphasis in Phase 2 is on effectiveness. This phase aims to obtain preliminary data on whether the drug works in people who have a certain disease or condition. For controlled trials, patients receiving the drug are compared with similar patients receiving a different treatment–usually an inactive substance (placebo), or a different drug. Safety continues to be evaluated, and short-term side effects are studied. Typically, the number of subjects in Phase 2 studies ranges from a few dozen to about 300.

At the end of Phase 2, the FDA and sponsors try to come to an agreement on how large-scale studies in Phase 3 should be done. How often the FDA meets with a sponsor varies, but this is one of two most common meeting points prior to submission of a new drug application. The other most common time is pre-NDA–right before a new drug application is submitted.
Phase 3 studies begin if evidence of effectiveness is shown in Phase 2. These studies gather more information about safety and effectiveness, studying different populations and different dosages and using the drug in combination with other drugs. The number of subjects usually ranges from several hundred to about 3,000 people.

Postmarket requirement and commitment studies are required of or agreed to by a sponsor, and are conducted after the FDA has approved a product for marketing. The FDA uses postmarket requirement and commitment studies to gather additional information about a product’s safety, efficacy, or optimal use.

New Drug Application (NDA)–This is the formal step a drug sponsor takes to ask that the FDA consider approving a new drug for marketing in the United States. An NDA includes all animal and human data and analyses of the data, as well as information about how the drug behaves in the body and how it is manufactured

When an NDA comes in, the FDA has 60 days to decide whether to file it so that it can be reviewed. The FDA can refuse to file an application that is incomplete. For example, some required studies may be missing. In accordance with the Prescription Drug User Fee Act (PDUFA), the FDA’s Center for Drug Evaluation and Research (CDER) expects to review and act on at least 90 percent of NDAs for standard drugs no later than 10 months after the applications are received. The review goal is six months for priority drugs. (See “The Role of User Fees.”)
“It’s the clinical trials that take so long–usually several years,” says Sandra Kweder, M.D., deputy director of the Office of New Drugs in the CDER. “The emphasis on speed for FDA mostly relates to review time and timelines of being able to meet with sponsors during a drug’s development,” she says.
Drug Approval Process Infographic
Drug Review Steps Simplified
- Preclinical (animal) testing.
- An investigational new drug application (IND) outlines what the sponsor of a new drug proposes for human testing in clinical trials.
- Phase 1 studies (typically involve 20 to 80 people).
- Phase 2 studies (typically involve a few dozen to about 300 people).
- Phase 3 studies (typically involve several hundred to about 3,000 people).
- The pre-NDA period, just before a new drug application (NDA) is submitted. A common time for the FDA and drug sponsors to meet.
- Submission of an NDA is the formal step asking the FDA to consider a drug for marketing approval.
- After an NDA is received, the FDA has 60 days to decide whether to file it so it can be reviewed.
- If the FDA files the NDA, an FDA review team is assigned to evaluate the sponsor’s research on the drug’s safety and effectiveness.
- The FDA reviews information that goes on a drug’s professional labeling (information on how to use the drug).
- The FDA inspects the facilities where the drug will be manufactured as part of the approval process.
- FDA reviewers will approve the application or issue a complete response letter.
Supplemental Information About the Drug Approval Process
Reviewing Applications
Though FDA reviewers are involved with a drug’s development throughout the IND stage, the official review time is the length of time it takes to review a new drug application and issue an action letter, an official statement informing a drug sponsor of the agency’s decision.
Once a new drug application is filed, an FDA review team–medical doctors, chemists, statisticians, microbiologists, pharmacologists, and other experts–evaluates whether the studies the sponsor submitted show that the drug is safe and effective for its proposed use. No drug is absolutely safe; all drugs have side effects. “Safe” in this sense means that the benefits of the drug appear to outweigh the known risks.
The review team analyzes study results and looks for possible issues with the application, such as weaknesses of the study design or analyses. Reviewers determine whether they agree with the sponsor’s results and conclusions, or whether they need any additional information to make a decision.
Each reviewer prepares a written evaluation containing conclusions and recommendations about the application. These evaluations are then considered by team leaders, division directors, and office directors, depending on the type of application.
Reviewers receive training that fosters consistency in drug reviews, and good review practices remain a high priority for the agency.
Sometimes, the FDA calls on advisory committees, who provide FDA with independent opinions and recommendations from outside experts on applications to market new drugs, and on FDA policies. Whether an advisory committee is needed depends on many things.
“Some considerations would be if it’s a drug that has significant questions, if it’s the first in its class, or the first for a given indication,” says Mark Goldberger, M.D., a former director of one of CDER’s drug review offices. “Generally, FDA takes the advice of advisory committees, but not always,” he says. “Their role is just that–to advise.”Accelerated Approval
Traditional approval requires that clinical benefit be shown before approval can be granted. Accelerated approval is given to some new drugs for serious and life-threatening illnesses that lack satisfactory treatments. This allows an NDA to be approved before measures of effectiveness that would usually be required for approval are available.
Instead, less traditional measures called surrogate endpoints are used to evaluate effectiveness. These are laboratory findings or signs that may not be a direct measurement of how a patient feels, functions, or survives, but are considered likely to predict benefit. For example, a surrogate endpoint could be the lowering of HIV blood levels for short periods of time with anti-retroviral drugs.
Gleevec (imatinib mesylate), an oral treatment for patients with a life-threatening form of cancer called chronic myeloid leukemia (CML), received accelerated approval. The drug was also approved under the FDA’s orphan drug program, which gives financial incentives to sponsors for manufacturing drugs that treat rare diseases. Gleevec blocks enzymes that play a role in cancer growth. The approval was based on results of three large Phase 2 studies, which showed the drug could substantially reduce the level of cancerous cells in the bone marrow and blood.
Most drugs to treat HIV have been approved under accelerated approval provisions, with the company required to continue its studies after the drug is on the market to confirm that its effects on virus levels are maintained and that it ultimately benefits the patient. Under accelerated approval rules, if studies don’t confirm the initial results, the FDA can withdraw the approval.
Because premarket review can’t catch all potential problems with a drug, the FDA continues to track approved drugs for adverse events through a postmarketing surveillance program.
Bumps in the Road
If the FDA decides that the benefits of a drug outweigh the known risks, the drug will receive approval and can be marketed in the United States. But if there are problems with an NDA or if more information is necessary to make that determination, the FDA may issue a complete response letter.
Common problems include unexpected safety issues that crop up or failure to demonstrate a drug’s effectiveness. A sponsor may need to conduct additional studies–perhaps studies of more people, different types of people, or for a longer period of time.
Manufacturing issues are also among the reasons that approval may be delayed or denied. Drugs must be manufactured in accordance with standards called good manufacturing practices, and the FDA inspects manufacturing facilities before a drug can be approved. If a facility isn’t ready for inspection, approval can be delayed. Any manufacturing deficiencies found need to be corrected before approval.
“Sometimes a company may make a certain amount of a drug for clinical trials. Then when they go to scale up, they may lose a supplier or end up with quality control issues that result in a product of different chemistry,” says Kweder. “Sponsors have to show us that the product that’s going to be marketed is the same product that they tested.”
John Jenkins, M.D., director of CDER’s Office of New Drugs, says, “It’s often a combination of problems that prevent approval.” Close communication with the FDA early on in a drug’s development reduces the chance that an application will have to go through more than one cycle of review, he says. “But it’s no guarantee.”
The FDA outlines the justification for its decision in a complete response letter to the drug sponsor and CDER gives the sponsor a chance to meet with agency officials to discuss the deficiencies. At that point, the sponsor can ask for a hearing, correct any deficiencies and submit new information, or withdraw the application.
The Role of User Fees
Since PDUFA was passed in 1992, more than 1,000 drugs and biologics have come to the market, including new medicines to treat cancer, AIDS, cardiovascular disease, and life-threatening infections. PDUFA has allowed the Food and Drug Administration to bring access to new drugs as fast or faster than anywhere in the world, while maintaining the same thorough review process.
Under PDUFA, drug companies agree to pay fees that boost FDA resources, and the FDA agrees to time goals for its review of new drug applications. Along with supporting increased staff, drug user fees help the FDA upgrade resources in information technology. The agency has moved toward an electronic submission and review environment, now accepting more electronic applications and archiving review documents electronically.
The goals set by PDUFA apply to the review of original new human drug and biological applications, resubmissions of original applications, and supplements to approved applications. The second phase of PDUFA, known as PDUFA II, was reauthorized in 1997 and extended the user fee program through September 2002. PDUFA III, which extended to Sept. 30, 2007, was reauthorized in June 2002.
PDUFA III allowed the FDA to spend some user fees to increase surveillance of the safety of medicines during their first two years on the market, or three years for potentially dangerous medications. It is during this initial period, when new medicines enter into wide use, that the agency is best able to identify and counter adverse side effects that did not appear during the clinical trials.
On September 27, 2007, President Bush signed into law the Food and Drug Administration Amendments Act of 2007 which includes the reauthorization and expansion of the Prescription Drug User Fee Act. The reauthorization of PDUFA will significantly broaden and upgrade the agency’s drug safety program, and facilitate more efficient development of safe and effective new medications for the American public.
In addition to setting time frames for review of applications, PDUFA sets goals to improve communication and sets goals for specific kinds of meetings between the FDA and drug sponsors. It also outlines how fast the FDA must respond to requests from sponsors. Throughout a drug’s development, the FDA advises sponsors on how to study certain classes of drugs, how to submit data, what kind of data are needed, and how clinical trials should be designed.
The Quality of Clinical Data
The Food and Drug Administration relies on data that sponsors submit to decide whether a drug should be approved. To protect the rights and welfare of people in clinical trials, and to verify the quality and integrity of data submitted, the FDA’s Division of Scientific Investigations (DSI) conducts inspections of clinical investigators’ study sites. DSI also reviews the records of institutional review boards to be sure they are fulfilling their role in patient protection.
“FDA investigators compare information that clinical investigators provided to sponsors on case report forms with information in source documents such as medical records and lab results,” says Carolyn Hommel, a consumer safety officer in DSI.
DSI seeks to determine such things as whether the study was conducted according to the investigational plan, whether all adverse events were recorded, and whether the subjects met the inclusion/exclusion criteria outlined in the study protocol.
At the conclusion of each inspection, FDA investigators prepare a report summarizing any deficiencies. In cases where they observe numerous or serious deviations, such as falsification of data, DSI classifies the inspection as “official action indicated” and sends a warning letter or Notice of Initiation of Disqualification Proceedings and Opportunity to Explain (NIDPOE) to the clinical investigator, specifying the deviations that were found.
The NIDPOE begins an administrative process to determine whether the clinical investigator should remain eligible to receive investigational products and conduct clinical studies.
CDER conducts about 300-400 clinical investigator inspections annually. About 3 percent are classified in this “official action indicated” category.
The FDA has established an independent Drug Safety Oversight Board (DSOB) to oversee the management of drug safety issues. The Board meets monthly and has representatives from three FDA Centers and five other federal government agencies. The board’s responsibilities include conducting timely and comprehensive evaluations of emerging drug safety issues, and ensuring that experts–both inside and outside of the FDA–give their perspectives to the agency. The first meeting of the DSOB was held in June 2005.
Once the review is complete, the NDA might be approved or rejected. If the drug is not approved, the applicant is given the reasons why and what information could be provided to make the application acceptable. Sometimes the FDA makes a tentative approval recommendation, requesting that a minor deficiency or labeling issue be corrected before final approval. Once a drug is approved, it can be marketed.
Some approvals contain conditions that must be met after initial marketing, such as conducting additional clinical studies. For example, the FDA might request a postmarketing, or phase 4, study to examine the risks and benefits of the new drug in a different population or to conduct special monitoring in a high-risk population. Alternatively, a phase 4 study might be initiated by the sponsor to assess such issues as the longer term effects of drug exposure, to optimize the dose for marketing, to evaluate the effects in pediatric patients, or to examine the effectiveness of the drug for additional indications. Postmarketing surveillance is important, because even the most well-designed phase 3 studies might not uncover every problem that could become apparent once a product is widely used. Furthermore, the new product might be more widely used by groups that might not have been well studied in the clinical trials, such as elderly patients. A crucial element in this process is that physicians report any untoward complications. The FDA has set up a medical reporting program called Medwatch to track serious adverse events (1-800-FDA-1088). The manufacturer must report adverse drug reactions at quarterly intervals for the first 3 years after approval, including a special report for any serious and unexpected adverse reactions.
Recent Developments in Drug Approval
The Food and Drug Administration Modernization Act of 1997 (FDAMA) extended the use of user fees and focused on streamlining the drug approval process. In 1999, the 35 drugs approved by the FDA were reviewed in an average of 12.6 months, slightly more than the 12-month goal set by PDUFA. This act also increased patient access to experimental drugs and facilitated an accelerated review of important new medications. The law ended the ban on disseminating information to providers about non-FDA-approved uses of medications. A manufacturer can now provide peer-reviewed journal articles about an off-label indication of a product if the company commits to filing a supplemental application to establish the use of the unapproved indication. As part of this process, the company must still conduct its own phase 4 study. As a condition for an accelerated approval, the FDA can require the sponsor to carry out postmarketing studies to confirm a clinical benefit and product safety. Critics contend the 1997 act compromises public safety by lowering the standard of approval. Within a year after the law was passed, several drugs were removed from the market. Among these medications were mibefradil for hypertension, dexfenfluramine for morbid obesity, the antihistamine terfenadine, and bromfenac sodium for pain. More recently, additional drugs including troglitazone were removed from the market. Although the increase in recalls might reflect the dramatic increase in drugs approved and launched, others argue that several safety questions were ignored. Another concern was that many withdrawn drugs were me-too drugs which did not represent a noteworthy advance in therapy. Persons critical of the FDA believe changes in the approval process, such as allowing some new drugs to be approved based on only a single clinical trial, expanded use of accelerated approvals, and the use of surrogate end points, have created a dangerous situation. Proponents of the changes in the approval process argue that there is no evidence of increased risk from the legislative changes, and that these changes improve access to cancer patients and those with debilitating disease who were previously denied critical and lifesaving medications.
New drugs are an important part of modern medicine. Just a few decades ago, a disease such as peptic ulcers was a frequent indication for major surgery. The advent of new pharmacologic treatments has dramatically reduced the serious complications of peptic ulcer disease. Likewise, thanks to many new antiviral medications, the outlook for HIV-infected patients has improved dramatically. It is important that physicians understand the process of approving these new medications. Understanding the process can promote innovation, help physicians assess new products, underline the importance of reporting adverse drug events, and provide physicians with the information to educate patients about participating in a clinical trial.



73,971 total views, 5 views today
