Crystallization













Crystallization is the (natural or artificial) process by which a solid forms, where the atoms or molecules are highly organized into a structure known as a crystal. Some of the ways by which crystals form are precipitating from a solution, freezing, or more rarely depositiondirectly from a gas. Attributes of the resulting crystal depend largely on factors such as temperature, air pressure, and in the case of liquid crystals, time of fluid evaporation.
Crystallization occurs in two major steps. The first is nucleation, the appearance of a crystalline phase from either a supercooled liquid or a supersaturated solvent. The second step is known as crystal growth, which is the increase in the size of particles and leads to a crystal state. An important feature of this step is that loose particles form layers at the crystal’s surface lodge themselves into open inconsistencies such as pores, cracks, etc.
The majority of minerals and organic molecules crystallize easily, and the resulting crystals are generally of good quality, i.e. without visible defects. However, larger biochemical particles, like proteins, are often difficult to crystallize. The ease with which molecules will crystallize strongly depends on the intensity of either atomic forces (in the case of mineral substances), intermolecular forces (organic and biochemical substances) or intramolecular forces (biochemical substances).
Crystallization is also a chemical solid–liquid separation technique, in which mass transfer of a solute from the liquid solution to a pure solid crystalline phase occurs. In chemical engineering, crystallization occurs in a crystallizer. Crystallization is therefore related to precipitation, although the result is not amorphous or disordered, but a crystal.
The design of a successful crystallization process depends on choosing process parameters that will produce crystals of the required purity and yield, that can be isolated, filtered, and dried easily. Process parameters such as cooling rate, solvent composition, and agitation rate directly impact crystallization behavior. Scientists are tasked with understanding how these parameters will influence the outcome of the crystallization process. Often, process parameters for crystallization are chosen based on previous experience, and the outcome is determined by careful analysis of offline analytical data, such as particle size analysis, XRPD, or microscopy. This approach is common, but neglects to consider that crystallization occurs through a sequence of interdependent mechanisms which all contribute to the final outcome, and are each uniquely influenced by the choice of process parameters.
Crystal nucleation and growth, phase separation, breakage, agglomeration, and polymorph transformations can occur separately, but also simultaneously, and are influenced by process parameters in unique ways. This convolution of mechanisms can mask the true role process parameters play in determining the outcome of a crystallization process, and make crystallization process design a particular challenge for scientists. In the absence of mechanistic understanding for crystallization processes, scientists must often rely on trial-and-error to adjust process parameters and optimize yield, purity, and particle size. This can be a time-consuming task and is one that rarely delivers crystals that can be isolated, filtered, and dried in a facile manner.
In this series of articles, the most common crystallization mechanisms are described alongside strategies to optimize them. The complete guide to crystallization mechanisms can be downloaded here.
What is Nucleation?
Nucleation occurs when solute molecules assemble in a supersaturated solution and reach a critical size. Primary nucleation occurs when nuclei appear from a solution directly and secondary nucleation occurs when nulcei appear in the presence of solids. Nucleation is important to understand because the number and size of nuclei formed can have a dominant influence on the final outcome of the crystallization process. High nucleation rates can lead to excessive fines and a bimodal crystal population which can make product isolation, filtration, and further processing difficult.

Considerations for Control
The nucleation rate is dependent on the molecule being crystallized but can be manipulated by considering the solvent type, controlling the supersaturation level, and evaluating the role of impurities and mixing during crystallization design. Seeding is a common strategy deployed to control primary nucleation. Effective seeding can initiate nucleation at a consistent point, and by choosing the seed size and seed loading the nucleation rate can be controlled.
Secondary nucleation often occurs during a crystallization process when supersaturation increases above a critical limit. This can occur when cooling is too fast or when anti-solvent is added quickly in an effort to increase yield. Secondary nucleation is particularly critical to understand and control because it can suddenly appear during scale-up when process parameters are controlled with less precision compared to the lab
Key Crystallization Definitions
Crystallization
Crystallization is a process whereby solid crystals are formed from another phase, typically a liquid solution or melt.
Crystal
Crystal is a solid particle in which the constituent molecules, atoms, or ions are arranged in some fixed and rigid, repeating three-dimensional pattern or lattice.
Precipitation
Precipitation is another word for crystallization but is most often used when crystallization occurs very quickly through a chemical reaction.
Solubility
Solubility is a measure of the amount of solute that can be dissolved in a given solvent at a given temperature
Saturated Solution
At a given temperature, there is a maximum amount of solute that can be dissolved in the solvent. At this point the solution is saturated. The quantity of solute dissolved at this point is the solubility.
Supersaturation
Supersaturation is the difference between the actual solute concentration and the equilibrium solute concentration at a given temperature.

Process

Time-lapse of growth of a citric acidcrystal. The video covers an area of 2.0 by 1.5 mm and was captured over 7.2 min.
The crystallization process consists of two major events, nucleation and crystal growth which are driven by thermodynamic properties as well as chemical properties. In crystallization Nucleation is the step where the solute molecules or atoms dispersed in the solvent start to gather into clusters, on the microscopic scale (elevating solute concentration in a small region), that become stable under the current operating conditions. These stable clusters constitute the nuclei. Therefore, the clusters need to reach a critical size in order to become stable nuclei. Such critical size is dictated by many different factors (temperature, supersaturation, etc.). It is at the stage of nucleation that the atoms or molecules arrange in a defined and periodic manner that defines the crystal structure — note that “crystal structure” is a special term that refers to the relative arrangement of the atoms or molecules, not the macroscopic properties of the crystal (size and shape), although those are a result of the internal crystal structure.
The crystal growth is the subsequent size increase of the nuclei that succeed in achieving the critical cluster size. Crystal growth is a dynamic process occurring in equilibrium where solute molecules or atoms precipitate out of solution, and dissolve back into solution. Supersaturation is one of the driving forces of crystallization, as the solubility of a species is an equilibrium process quantified by Ksp. Depending upon the conditions, either nucleation or growth may be predominant over the other, dictating crystal size.
Many compounds have the ability to crystallize with some having different crystal structures, a phenomenon called polymorphism. Each polymorph is in fact a different thermodynamic solid state and crystal polymorphs of the same compound exhibit different physical properties, such as dissolution rate, shape (angles between facets and facet growth rates), melting point, etc. For this reason, polymorphism is of major importance in industrial manufacture of crystalline products. Additionally, crystal phases can sometimes be interconverted by varying factors such as temperature.
In nature

Snowflakes are a very well-known example, where subtle differences in crystal growth conditions result in different geometries.

Crystallized honey
There are many examples of natural process that involve crystallization.
Geological time scale process examples include:
- Natural (mineral) crystal formation (see also gemstone);
- Stalactite/stalagmite, rings formation.
Human time scale process examples include:
- Snow flakes formation;
- Honey crystallization (nearly all types of honey crystallize).
Methods
Crystal formation can be divided into two types, where the first type of crystals are composed of a cation and anion, also known as a salt, such as sodium acetate. The second type of crystals are composed of uncharged species, for example menthol.[1]
Crystal formation can be achieved by various methods, such as: cooling, evaporation, addition of a second solvent to reduce the solubility of the solute (technique known as antisolvent or drown-out), solvent layering, sublimation, changing the cation or anion, as well as other methods.
The formation of a supersaturated solution does not guarantee crystal formation, and often a seed crystal or scratching the glass is required to form nucleation sites.
A typical laboratory technique for crystal formation is to dissolve the solid in a solution in which it is partially soluble, usually at high temperatures to obtain supersaturation. The hot mixture is then filtered to remove any insoluble impurities. The filtrate is allowed to slowly cool. Crystals that form are then filtered and washed with a solvent in which they are not soluble, but is miscible with the mother liquor. The process is then repeated to increase the purity in a technique known as recrystallization.
For biological molecules in which the solvent channels continue to be present to retain the three dimensional structure intact, microbatch[2]crystallization under oil and vapor diffusion[3] methods have been the common methods.
Typical equipment
Equipment for the main industrial processes for crystallization.
- Tank crystallizers. Tank crystallization is an old method still used in some specialized cases. Saturated solutions, in tank crystallization, are allowed to cool in open tanks. After a period of time the mother liquor is drained and the crystals removed. Nucleation and size of crystals are difficult to control. Typically, labor costs are very high
Thermodynamic view

Low-temperature SEMmagnification series for a snow crystal. The crystals are captured, stored, and sputter-coated with platinum at cryo-temperatures for imaging.
The crystallization process appears to violate the second principle of thermodynamics. Whereas most processes that yield more orderly results are achieved by applying heat, crystals usually form at lower temperatures—especially by supercooling. However, due to the release of the heat of fusion during crystallization, the entropy of the universe increases, thus this principle remains unaltered.
The molecules within a pure, perfect crystal, when heated by an external source, will become liquid. This occurs at a sharply defined temperature (different for each type of crystal). As it liquifies, the complicated architecture of the crystal collapses. Melting occurs because the entropy (S) gain in the system by spatial randomization of the molecules has overcome the enthalpy (H) loss due to breaking the crystal packing forces:
- {\displaystyle T(S_{\text{liquid}}-S_{\text{solid}})>H_{\text{liquid}}-H_{\text{solid}},}
- {\displaystyle G_{\text{liquid}}<G_{\text{solid}}.}


Regarding crystals, there are no exceptions to this rule. Similarly, when the molten crystal is cooled, the molecules will return to their crystalline form once the temperature falls beyond the turning point. This is because the thermal randomization of the surroundings compensates for the loss of entropy that results from the reordering of molecules within the system. Such liquids that crystallize on cooling are the exception rather than the rule.
The nature of a crystallization process is governed by both thermodynamic and kinetic factors, which can make it highly variable and difficult to control. Factors such as impurity level, mixing regime, vessel design, and cooling profile can have a major impact on the size, number, and shape of crystals produced.
Dynamics
As mentioned above, a crystal is formed following a well-defined pattern, or structure, dictated by forces acting at the molecular level. As a consequence, during its formation process the crystal is in an environment where the solute concentration reaches a certain critical value, before changing status. Solid formation, impossible below the solubility threshold at the given temperature and pressure conditions, may then take place at a concentration higher than the theoretical solubility level. The difference between the actual value of the solute concentration at the crystallization limit and the theoretical (static) solubility threshold is called supersaturation and is a fundamental factor in crystallization.
Nucleation
Nucleation is the initiation of a phase change in a small region, such as the formation of a solid crystal from a liquid solution. It is a consequence of rapid local fluctuations on a molecular scale in a homogeneous phase that is in a state of metastable equilibrium. Total nucleation is the sum effect of two categories of nucleation – primary and secondary.
Primary nucleation
Primary nucleation is the initial formation of a crystal where there are no other crystals present or where, if there are crystals present in the system, they do not have any influence on the process. This can occur in two conditions. The first is homogeneous nucleation, which is nucleation that is not influenced in any way by solids. These solids include the walls of the crystallizer vessel and particles of any foreign substance. The second category, then, is heterogeneous nucleation. This occurs when solid particles of foreign substances cause an increase in the rate of nucleation that would otherwise not be seen without the existence of these foreign particles. Homogeneous nucleation rarely occurs in practice due to the high energy necessary to begin nucleation without a solid surface to catalyse the nucleation.
Primary nucleation (both homogeneous and heterogeneous) has been modelled with the following:[4]
- {\displaystyle B={\dfrac {dN}{dt}}=k_{n}(c-c^{*})^{n},}

where
- B is the number of nuclei formed per unit volume per unit time,
- N is the number of nuclei per unit volume,
- kn is a rate constant,
- c is the instantaneous solute concentration,
- c* is the solute concentration at saturation,
- (c − c*) is also known as supersaturation,
- n is an empirical exponent that can be as large as 10, but generally ranges between 3 and 4.
Secondary nucleation
Secondary nucleation is the formation of nuclei attributable to the influence of the existing microscopic crystals in the magma.[5] Simply put, secondary nucleation is when crystal growth is initiated with contact of other existing crystals or “seeds”.[6] The first type of known secondary crystallization is attributable to fluid shear, the other due to collisions between already existing crystals with either a solid surface of the crystallizer or with other crystals themselves. Fluid-shear nucleation occurs when liquid travels across a crystal at a high speed, sweeping away nuclei that would otherwise be incorporated into a crystal, causing the swept-away nuclei to become new crystals. Contact nucleation has been found to be the most effective and common method for nucleation. The benefits include the following:[5]
- Low kinetic order and rate-proportional to supersaturation, allowing easy control without unstable operation.
- Occurs at low supersaturation, where growth rate is optimal for good quality.
- Low necessary energy at which crystals strike avoids the breaking of existing crystals into new crystals.
- The quantitative fundamentals have already been isolated and are being incorporated into practice.
The following model, although somewhat simplified, is often used to model secondary nucleation:[4]
- {\displaystyle B={\dfrac {dN}{dt}}=k_{1}M_{T}^{j}(c-c^{*})^{b},}

where
- k1 is a rate constant,
- MT is the suspension density,
- j is an empirical exponent that can range up to 1.5, but is generally 1,
- b is an empirical exponent that can range up to 5, but is generally 2.

Growth
Once the first small crystal, the nucleus, forms it acts as a convergence point (if unstable due to supersaturation) for molecules of solute touching – or adjacent to – the crystal so that it increases its own dimension in successive layers. The pattern of growth resembles the rings of an onion, as shown in the picture, where each colour indicates the same mass of solute; this mass creates increasingly thin layers due to the increasing surface area of the growing crystal. The supersaturated solute mass the original nucleus may capture in a time unit is called the growth rate expressed in kg/(m2*h), and is a constant specific to the process. Growth rate is influenced by several physical factors, such as surface tension of solution, pressure, temperature, relative crystal velocity in the solution, Reynolds number, and so forth.
The main values to control are therefore:
- Supersaturation value, as an index of the quantity of solute available for the growth of the crystal;
- Total crystal surface in unit fluid mass, as an index of the capability of the solute to fix onto the crystal;
- Retention time, as an index of the probability of a molecule of solute to come into contact with an existing crystal;
- Flow pattern, again as an index of the probability of a molecule of solute to come into contact with an existing crystal (higher in laminar flow, lower in turbulent flow, but the reverse applies to the probability of contact).
The first value is a consequence of the physical characteristics of the solution, while the others define a difference between a well- and poorly designed crystallizer.
Size distribution
Main crystallization processes[]The appearance and size range of a crystalline product is extremely important in crystallization. If further processing of the crystals is desired, large crystals with uniform size are important for washing, filtering, transportation, and storage, because large crystals are easier to filter out of a solution than small crystals. Also, larger crystals have a smaller surface area to volume ratio, leading to a higher purity. This higher purity is due to less retention of mother liquor which contains impurities, and a smaller loss of yield when the crystals are washed to remove the mother liquor. The theoretical crystal size distribution can be estimated as a function of operating conditions with a fairly complicated mathematical process called population balance theory (using population balance equations).

Some of the important factors influencing solubility are:
- Concentration
- Temperature
- Polarity
- Ionic strength
So one may identify two main families of crystallization processes:
- Cooling crystallization
- Evaporative crystallization
This division is not really clear-cut, since hybrid systems exist, where cooling is performed through evaporation, thus obtaining at the same time a concentration of the solution.
A crystallization process often referred to in chemical engineering is the fractional crystallization. This is not a different process, rather a special application of one (or both) of the above.
Cooling crystallization
Application
Most chemical compounds, dissolved in most solvents, show the so-called direct solubility that is, the solubility threshold increases with temperature.

Solubility of the system Na2SO4 – H2O
So, whenever the conditions are favourable, crystal formation results from simply cooling the solution. Here cooling is a relative term: austenite crystals in a steel form well above 1000 °C. An example of this crystallization process is the production of Glauber’s salt, a crystalline form of sodium sulfate. In the diagram, where equilibrium temperature is on the x-axis and equilibrium concentration (as mass percent of solute in saturated solution) in y-axis, it is clear that sulfate solubility quickly decreases below 32.5 °C. Assuming a saturated solution at 30 °C, by cooling it to 0 °C (note that this is possible thanks to the freezing-point depression), the precipitation of a mass of sulfate occurs corresponding to the change in solubility from 29% (equilibrium value at 30 °C) to approximately 4.5% (at 0 °C) – actually a larger crystal mass is precipitated, since sulfate entrains hydration water, and this has the side effect of increasing the final concentration.
There are limitations in the use of cooling crystallization:
- Many solutes precipitate in hydrate form at low temperatures: in the previous example this is acceptable, and even useful, but it may be detrimental when, for example, the mass of water of hydration to reach a stable hydrate crystallization form is more than the available water: a single block of hydrate solute will be formed – this occurs in the case of calcium chloride);
- Maximum supersaturation will take place in the coldest points. These may be the heat exchanger tubes which are sensitive to scaling, and heat exchange may be greatly reduced or discontinued;
- A decrease in temperature usually implies an increase of the viscosity of a solution. Too high a viscosity may give hydraulic problems, and the laminar flow thus created may affect the crystallization dynamics.
- It is not applicable to compounds having reverse solubility, a term to indicate that solubility increases with temperature decrease (an example occurs with sodium sulfate where solubility is reversed above 32.5 °C).
Cooling crystallizers

Vertical cooling crystallizer in a beet sugar factory
The simplest cooling crystallizers are tanks provided with a mixer for internal circulation, where temperature decrease is obtained by heat exchange with an intermediate fluid circulating in a jacket. These simple machines are used in batch processes, as in processing of pharmaceuticals and are prone to scaling. Batch processes normally provide a relatively variable quality of product along the batch.
The Swenson-Walker crystallizer is a model, specifically conceived by Swenson Co. around 1920, having a semicylindric horizontal hollow trough in which a hollow screw conveyor or some hollow discs, in which a refrigerating fluid is circulated, plunge during rotation on a longitudinal axis. The refrigerating fluid is sometimes also circulated in a jacket around the trough. Crystals precipitate on the cold surfaces of the screw/discs, from which they are removed by scrapers and settle on the bottom of the trough. The screw, if provided, pushes the slurry towards a discharge port.
A common practice is to cool the solutions by flash evaporation: when a liquid at a given T0 temperature is transferred in a chamber at a pressure P1 such that the liquid saturation temperature T1 at P1 is lower than T0, the liquid will release heat according to the temperature difference and a quantity of solvent, whose total latent heat of vaporization equals the difference in enthalpy. In simple words, the liquid is cooled by evaporating a part of it.
In the sugar industry, vertical cooling crystallizers are used to exhaust the molasses in the last crystallization stage downstream of vacuum pans, prior to centrifugation. The massecuite enters the crystallizers at the top, and cooling water is pumped through pipes in counterflow.
Evaporative crystallization
Another option is to obtain, at an approximately constant temperature, the precipitation of the crystals by increasing the solute concentration above the solubility threshold. To obtain this, the solute/solvent mass ratio is increased using the technique of evaporation. This process is insensitive to change in temperature (as long as hydration state remains unchanged).
All considerations on control of crystallization parameters are the same as for the cooling models.
Evaporative crystallizers
Most industrial crystallizers are of the evaporative type, such as the very large sodium chloride and sucrose units, whose production accounts for more than 50% of the total world production of crystals. The most common type is the forced circulation (FC) model (see evaporator). A pumping device (a pump or an axial flow mixer) keeps the crystal slurry in homogeneous suspension throughout the tank, including the exchange surfaces; by controlling pump flow, control of the contact time of the crystal mass with the supersaturated solution is achieved, together with reasonable velocities at the exchange surfaces. The Oslo, mentioned above, is a refining of the evaporative forced circulation crystallizer, now equipped with a large crystals settling zone to increase the retention time (usually low in the FC) and to roughly separate heavy slurry zones from clear liquid. Evaporative crystallizers tend to yield larger average crystal size and narrows the crystal size distribution curve.[7]
DTB crystallizer


Schematic of DTB
Whichever the form of the crystallizer, to achieve an effective process control it is important to control the retention time and the crystal mass, to obtain the optimum conditions in terms of crystal specific surface and the fastest possible growth. This is achieved by a separation – to put it simply – of the crystals from the liquid mass, in order to manage the two flows in a different way. The practical way is to perform a gravity settling to be able to extract (and possibly recycle separately) the (almost) clear liquid, while managing the mass flow around the crystallizer to obtain a precise slurry density elsewhere. A typical example is the DTB (Draft Tube and Baffle) crystallizer, an idea of Richard Chisum Bennett (a Swenson engineer and later President of Swenson) at the end of the 1950s. The DTB crystallizer (see images) has an internal circulator, typically an axial flow mixer – yellow – pushing upwards in a draft tube while outside the crystallizer there is a settling area in an annulus; in it the exhaust solution moves upwards at a very low velocity, so that large crystals settle – and return to the main circulation – while only the fines, below a given grain size are extracted and eventually destroyed by increasing or decreasing temperature, thus creating additional supersaturation. A quasi-perfect control of all parameters is achieved as DTF crystallizers offer superior control over crystal size and characteristics.[8] This crystallizer, and the derivative models (Krystal, CSC, etc.) could be the ultimate solution if not for a major limitation in the evaporative capacity, due to the limited diameter of the vapour head and the relatively low external circulation not allowing large amounts of energy to be supplied to the system.
Crystallization and Polymorphism
The physical form of an active pharmaceutical ingredient (API) is critical for successful drug development. The final step in the production of an API is typically crystallization, in which impurities are rejected in the mother liquor and the desired solid form is produced. This form should be stable and have suitable solubility and handling characteristics for downstream processing activities such as formulation.

The physical form of an active pharmaceutical ingredient (API) is critical for successful drug development. The final step in the production of an API is typically crystallization, in which impurities are rejected in the mother liquor and the desired solid form is produced. This form should be stable and have suitable solubility and handling characteristics for downstream processing activities such as formulation.
Obtaining the right form, however, is not always a straightforward process. For example, axitinib (trade name Inlyta, 1 in Figure 1), a Pfizer’s breast cancer drug, can form 66 different solvates and 5 anhydrous polymorphs. (Campeta, A. M. et al. J. Pharm. Sci. DOI: 10.1002/jps.22230; Chekal, B. P., et al. Org. Process Res. Dev. DOI: 10.1021/op9001559)
The appearance of new solid forms (see box) during development can be a real problem if they are thermodynamically more stable and less soluble than the desired form.
Several case studies on this subject that illustrate some of the problems that can arise recently were published in Organic Process Research and Development. These articles will likely appear in a special issue on crystallization and polymorphism.
Common Solid Forms
- Hydrate: Water is an integral part of the crystal lattice.
- Solvate: A solvent molecule is an integral part of the crystal lattice.
- Polymorphs: Multiple forms of a molecule that have different crystal structures.
Raltegravir prodrug
Merck markets raltegravir (Isentress, 2 in Figure 2) as a treatment for HIV/AIDS infection. The company is currently developing an acetal carbonate prodrug (MK-8970, 3) of 2, which has better absorption and physicochemical properties. Merck researchers have identified two solid forms of 3.

Merck markets raltegravir (Isentress, 2 in Figure 2) as a treatment for HIV/AIDS infection. The company is currently developing an acetal carbonate prodrug (MK-8970, 3) of 2, which has better absorption and physicochemical properties. Merck researchers have identified two solid forms of 3.
Both forms are anhydrous and are true racemates. Differential scanning calorimetry studies showed that the two forms are enantiotropically related, with a transition temperature estimated at 116.9 ºC. This means that form 2 is more stable below 117 ºC, whereas form 1 is more stable above 117 ºC.
During lab trials, an unexpected peak appeared in the X-ray powder diffraction (XRPD) pattern of some samples of 3. The peak was identified as an imidate impurity present at 1–2%; it has a very intense peak at exactly the right place in the XRPD pattern (bottom trace in Figure 3). A solvent screen showed that the imidate is less soluble than MK-8970 in ethyl acetate, the original solvent. The chemists found that Isopropyl acetate is a better solvent for obtaining pure form 2. (Shultz, C. S., et al. Org. Process Res. Dev.DOI: 10/1021/acs.oprd.5b00129)

A new form appears

AMG 579 (4 in Figure 4) is a potent and selective phosphodiesterase 10 inhibitor that Amgen is developing for treating schizophrenia. During lead optimization, Amgen researchers produced six batches of 4, all of which had the same crystalline form. Scaling up the crystallization during development required a longer cooling period, which resulted in the isolation of a new polymorph. A subsequent screen resulted in 303 crystalline samples from 384 crystallization conditions, but no forms other than forms 1 and 2 were found.
As with prodrug 3 discussed above, forms 1 and 2 are enantiotropically related. Both forms have many similar attributes (e.g., crystallinity, high melting point, lack of hygroscopicity, stability, and in vivo performance). But form 2 is much easier to produce in a manufacturing setting, and it was chosen for clinical development. (Kiang, Y.-H., et al. Org. Process Res. Dev.DOI:10.1021/acs.oprd.5b00031)
A new hydrated form

Organic compounds can also crystallize as solvates or hydrates; and, as with other solid forms, understanding the solid state “landscape” is critical to success. Amgen’s developmental API AMG A (5 in Figure 5) has one anhydrous form (form A) and four solvates. After 10 batches of form A were reliably produced, a new hydrated form appeared when a change was made to the seed preparation method during the good manufacturing practice project.
During this phase of development, seeds were sonicated for ≈20 min rather than being slurried for 1–2 h. This gave a serendipitous result because 40–100-g amounts of the hydrated form have a much shorter filtration time (1–2 min) than form A (10 h).
Figure 6 is a polarized light microscopy (PLM) image of form A that was produced from the original seeding protocol. Figure 7 shows PLM images of the seed (left) and final cake produced when the seed was prepared with sonication.


The new form is a stable hemihydrate; it was selected for further development. Water activity studies revealed that the hemihydrate form is more stable in propanol–water mixtures when the water content is >6.2 wt%. (Nagapudi, K., et al. Org. Process Res. Dev. DOI: 10.1021/acs.oprd.5b00030)
Computation is key
The complexity of the solid-state landscape seems set to continue for the foreseeable future. On occasion, new forms appear late in development and give headaches to chemists, chemical engineers, and formulators. Modern analytical techniques help, but predictive computational methods are most likely to improve our understanding of polymorphism.
What Is Crystallization?

Crystallization is the process of atoms or molecules arranging into a well-defined, rigid crystal lattice in order to minimize their energetic state. The smallest entity of crystal lattice is called a unit cell, which can accept atoms or molecules to grow a macroscopic crystal. During crystallization, atoms and molecules bind together with well-defined angles to form a characteristic crystal shape with smooth surfaces and facets. Although crystallization can occur in nature, crystallization also has a broad industrial application as a separation and purification step in the pharmaceutical and chemical industries.

The choice of operating conditions during a crystallization process directly influences important product attributes such as crystal size, crystal shape and purity. By understanding the crystallization process and choosing the right process parameters, it is possible to repeatedly produce crystals of the correct size, shape, and purity while minimizing issues downstream such as long filtration times or inadequate drying.
Why is Crystallization Important?
Crystallization touches every aspect of our lives from the foods we eat and the medicines we take, to the fuels we use to power our communities. The majority of agrochemical and pharmaceutical products go through many crystallization steps during their development and manufacture. Key food ingredients, such as lactose and lysine, are manufactured using crystallization and the unwanted crystallization of gas hydrates in deep sea pipelines is a major safety concern for the petrochemical industry
Types of Crystallization
Crystallization occurs when the solubility of a solute in solution is reduced by some means. Common methods to reduce solubility include:
a. Cooling
b. Anti-Solvent Addition
c. Evaporation
d. Reaction (Precipitation)
The choice of crystallization method depends on the equipment available for crystallization, the objectives of the crystallization process and the solubility and stability of the solute in the chosen solvent.

Common Crystallization Challenges
Crystallization proceeds through a series of interdependent mechanisms that are each uniquely influenced by the choice of process parameters:
- Nucleation
- Growth
- Oiling Out
- Agglomeration
- Breakage
- Polymorph Transition
These mechanisms, which are often hidden form scientists, play a dominant role in defining the outcome of a crystallization process.

Crystallization Steps
- Choose an appropriate solvent. Common considerations included how much solute can be dissolved (solubility) and how practical the solvent is to handle (safety)
- Dissolve the product in the solvent by increasing the temperature until the last product molecule disappears. At this insoluble impurities may be filtered from the hot solution
- Reduce solubility via cooling, anti-solvent addition, evaporation or reaction. The solution will become supersaturated.
- Crystallize the product. As solubility is reduced a point is reached where crystals will nucleate and then grow. Highly pure product crystals should form and impurities should remain in solution.
- Allow the system to reach equilibrium after cooling (or another crystallization method stops).
- Filter and dry the purified product.
-
How to Design a Crystallization Process
The design of a crystallization process that will deliver pure crystals with an optimized yield and size, involves considering a number of important elements:
- Choose an appropriate solvent
- Screen for stability and unwanted polymorphs
- Determine growth and nucleation kinetics
- Define a seeding strategy
- Optimize cooling and anti-solvent profiles
- Understand the impact of mixing and scale
Common Crystallization Parameters & Transformations
While crystals have many important attributes the crystal size distribution probably has the greatest impact on the quality and effectiveness of the final product (and the process needed to deliver it). Crystal size and shape directly influence key steps downstream from the crystallizer, with filtration and drying performance being particularly susceptible to changes in these important attributes. Similarly, the final crystal size can also directly influence the quality of the final product. In a pharmaceutical compound, bioavailability and efficacy are often related to particle size with smaller particles often desired for their enhanced solubility and dissolution characteristics
Crystal size distribution can be optimized and controlled by carefully choosing the correct crystallization conditions and process parameters. Understanding how process parameters influence key transformations, such as nucleation, growth, and breakage, allow scientists to develop and manufacture crystals that will have the desired attributes and be efficient to bring to the market.
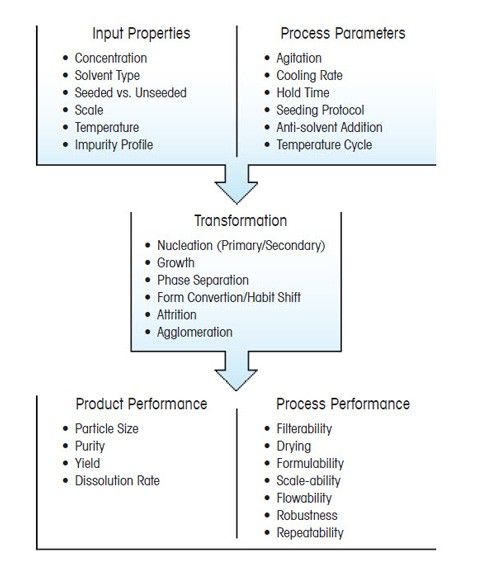
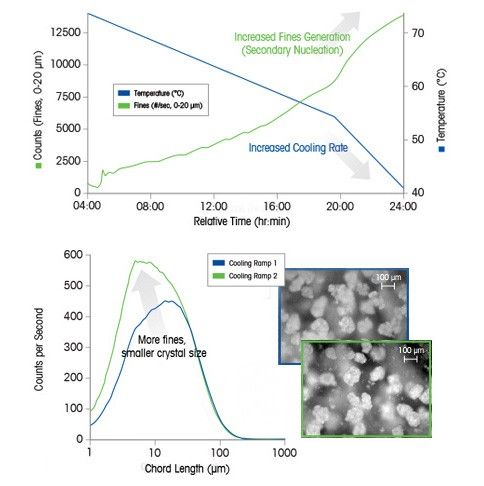
Case Study: Crystallization Cooling Rates
In this example, the cooling rate at the end of the batch induces secondary nucleation (monitored by ParticleTrack with FBRM technology) resulting in the formation of many fine particles – directly observed in real time using ParticleView with PVM technology.
An increase in cooling rate generates supersaturation faster – which is consumed by nucleation rather than growth. Careful control of cooling rate is critical to ensure the desired crystal size distribution specification can be achieved.
The crystal size distribution of ice plays a vital role in the taste and consistency of ice cream, with crystals smaller than 50 μm being better than crystals larger than 100 μm. For agrochemicals, it is vital to ensure that particles are small enough to be sprayed without blocking nozzles while large enough not to drift into neighboring fields.
While it is often a challenge to control crystal size distribution across scales, an opportunity exists to understand crystallization processes to deliver an optimized size and shape distribution that will ensure a cost effective process with the highest possible quality.
Technologies to Monitor, Optimize & Control
Crystallization unit operations offer the unique opportunity to target and control an optimized crystal size and shape distribution to:
- Reduce Filtration and Drying Times
- Avoid Storage, Transport and Shelf Life Issues
- Ensure a Consistent and Repeatable Process at Lower Costs
7 Key Crystallization Mechanisms
- Nucleation
- Growth
- Oiling Out
- Agglomeration
- Breakage
- Seeding
- Polymorph Transformation
Often, these mechanisms occur simultaneously making effective crystallization design a challenge. In the absence of mechanistic understanding, scientists rely on trial-and-error to adjust process parameters and optimize yield, purity, and crystal size. By understanding which mechanisms occur during crystallization, scientists can deploy strategies to deliver a crystal product with the desired properties.
Key Mechanistic Challenges
 |
Achieve consistent primary nucleation and avoid secondary nucleation events.2. Growth
Enhance growth over nucleation to increase crystal size.3. Oiling Out
Determine the phase diagram and avoid conditions where oiling out occurs.4. Agglomeration
Avoid agglomeration through solvent selection and parameter control.5. Breakage
Avoid unwanted breakage and addition and control desired wet-milling.6. Seeding
Optimize seeding protocol to deliver desired particle properties.7. Polymorph Transformation
Navigate the phase diagram to produce the desired form.
Busting a myth about mechanochemical crystallization


Although it may seem counterintuitive to put a compound into a ball mill to turn it into a crystalline form, the approach nonetheless works—and adding varying amounts of liquid can determine the crystal form that results, reports a team led by Bill Jones of the University of Cambridge (Cryst. Growth Des.2016, DOI: 10.1021/acs.cgd.6b00682).
Compounds of interest for materials and pharmaceuticals applications often crystallize into different forms, called polymorphs. Because polymorphs can have varying stability, solubility, and other properties, forming a specific polymorph can be critically important.
Chemists have long thought that using one particular liquid when crystallizing compounds via mechanochemical milling always yields one particular polymorph. Seeking to test that dogma, Jones and coworkers crystallized 200 mg of a 1:1 equimolar mixture of caffeine and anthranilic acid using a ball mill, adding from 10 to 100 μL of 15 different liquids.
Four liquids—acetonitrile, nitromethane, ethylene glycol, and 1,6-hexanediol—formed one polymorph each, regardless of the amount of liquid. The rest of the liquids yielded different polymorphs or mixtures, depending on liquid volume: 10 to 20 μL of ethanol formed polymorph II, for example, whereas 40 to 60 μL formed polymorph I. Additionally, 10 μL of 1-hexanol, 1-octanol, or 1-dodecanol formed polymorph III, a polymorph previously only prepared by desolvation.
Similar effects could occur for single-component crystals, the authors say. The mechanism behind the phenomenon remains to be determined; the authors suggest that it could be a result of thermodynamic stabilization of nanoparticles, different growth mechanisms of the polymorphs, or changes in the free-energy difference between polymorphs caused by milling conditions.
See also
- Abnormal grain growth
- Chiral resolution by crystallization
- Crystal habit
- Crystal structure
- Crystallite
- Fractional crystallization (chemistry)
- Igneous differentiation
- Laser heated pedestal growth
- Micro-pulling-down
- Protein crystallization
- Pumpable ice technology
- Quasicrystal
- Recrystallization (chemistry)
- Recrystallization (metallurgy)
- Seed crystal
- Single crystal
- Symplectite
- Vitrification
- X-ray crystallography
References
- Jump up^ Lin, Yibin (2008). “An Extensive Study of Protein Phase Diagram Modification:Increasing Macromolecular Crystallizability by Temperature Screening”. Crystal Growth & Design. 8 (12): 4277. doi:10.1021/cg800698p.
- Jump up^ Chayen, Blow (1992). “Microbatch crystallization under oil — a new technique allowing many small-volume crystallization trials”. Journal of Crystal Growth. 122 (1-4): 176-180. Bibcode:1992JCrGr.122..176C. doi:10.1016/0022-0248(92)90241-A.
- Jump up^ Benvenuti, Mangani (2007). “Crystallization of soluble proteins in vapor diffusion for x-ray crystallography”. Nature Protocols. 2: 1663. doi:10.1038/nprot.2007.198.
- ^ Jump up to:a b Tavare, N. S. (1995). Industrial Crystallization. Plenum Press, New York.
- ^ Jump up to:a b McCabe & Smith (2000). Unit Operations of Chemical Engineering. McGraw-Hill, New York.
- Jump up^ “Crystallization”. www.reciprocalnet.org. Archived from the original on 2016-11-27. Retrieved 2017-01-03.
- Jump up^ “Submerge Circulating Crystallizers – Thermal Kinetics Engineering, PLLC”. Thermal Kinetics Engineering, PLLC. Retrieved 2017-01-03.
- Jump up^ “Draft Tube Baffle (DTB) Crystallizer – Swenson Technology”. Swenson Technology. Archived from the original on 2016-09-25. Retrieved 2017-01-03.
Further reading
- A. Mersmann, Crystallization Technology Handbook (2001) CRC; 2nd ed. ISBN0-8247-0528-9
- Tine Arkenbout-de Vroome, Melt Crystallization Technology (1995) CRC ISBN1-56676-181-6
- “Small Molecule Crystallization” (PDF) at Illinois Institute of Technology website
- Glynn P.D. and Reardon E.J. (1990) “Solid-solution aqueous-solution equilibria: thermodynamic theory and representation”. Amer. J. Sci. 290, 164–201.
- Geankoplis, C.J. (2003) “Transport Processes and Separation Process Principles”. 4th Ed. Prentice-Hall Inc.
- S.J. Jancic, P.A.M. Grootscholten: “Industrial Crystallization”, Textbook, Delft University Press and Reidel Publishing Company, Delft, The Netherlands, 1984.
External links
Crystallization Publications
Discover a selection of crystallization publications below:
The seminal study on the nucleation of crystals from solution
Jaroslav Nývlt, Kinetics of nucleation in solutions, Journal of Crystal Growth, Volumes 3–4, 1968.
Excellent study on how crystals grow form solution
Crystal Growth Kinetics, Material Science and Engineering, Volume 65, Issue 1, July 1984.
An excellent description of the reasons solute-solvent systems exhibit oiling out instead of crystallization
Kiesow et al., Experimental investigation of oiling out during crystallization process, Journal of Crystal Growth, Volume 310, Issue 18, 2008.
Detailed examination of why agglomeration occurs during crystallization
Brunsteiner et al., Toward a Molecular Understanding of Crystal Agglomeration, Crystal Growth & Design, 2005, 5 (1), pp 3–16.
A study of breakage mechanisms during crystallization
Fasoli & Conti, Crystal breakage in a mixed suspension crystallizer, Volume 8, Issue8, 1973, Pages 931-946.
A great overview of how to design effective crystallization processes in the high value chemicals industry
Paul et al., Organic Crystallization Processes, Powder Technology, Volume 150, Issue 2, 2005.
Techniques to ensure the correct polymorph is crystallized every time
Kitamura, Strategies for Control of Crystallization of Polymorphs, CrystEngComm, 2009,11, 949-964.
////////////////
23,668 total views, 3 views today
