NINTEDANIB, BBIF 1120, Intedanib
Boehringer Ingelheim Corp
As a potential treatment for a range of different solid tumour types
CAS 656247-17-5
CAS 1377321-64-6 (nintedanib bisethanesulfonate)
CAS [656247-18-6] mono ethane sulfonate
3(Z)-[1-[4-[N-Methyl-N-[2-(4-methylpiperazin-1-yl)acetyl]amino]phenylamino]-1-phenylmethylene]-2-oxo-2,3-dihydro-1H-indole-6-carboxylic acid methyl ester
MW 539.62, MF C31 H33 N5 O4
Launched 2014 USA….Idiopathic pulmonary fibrosis
| chinese, japanese |
尼达尼布 ニンテダニブ |
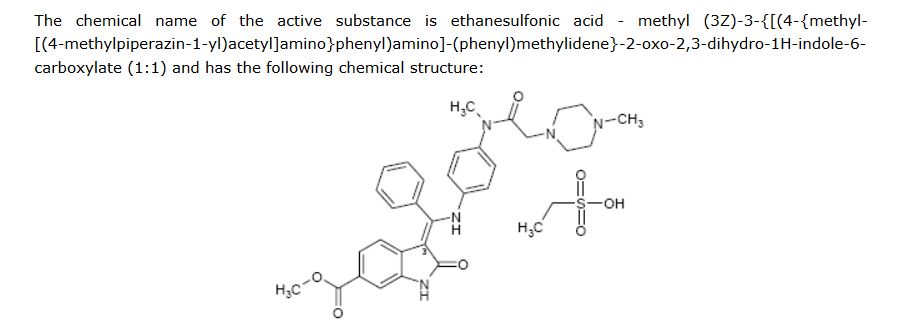
Ethanesulfonic acid – methyl (3Z)-3-{[(4-{methyl[(4-methyl-1-piperazinyl)acetyl]amino}phenyl)amino](phenyl)methylene}-2-oxo-6-indolinecarboxylate (1:1)
Nintedanib esylate
Cas 656247-18-6 [RN]
Methyl (3Z)-3-[({4-[N-methyl-2-(4-methylpiperazin-1-yl)acetamido]phenyl}amino)(phenyl)methylidene]-2-oxo-2,3-dihydro-1H-indole-6-carboxylate ethanesulfonate
Nintedanib esylate [USAN]
(3Z)-2,3-Dihydro-3-[[[4-[methyl[2-(4-methyl-1-piperazinyl)acetyl]amino]phenyl]amino]phenylmethylene]-2-oxo-1H-indole-6-carboxylic acid methyl ester ethanesulfonate
1H-Indole-6-carboxylic acid, 2,3dihydro-3-[[[4-[methyl[(4-methyl-1-piperazinyl)acetyl]amino]phenyl]amino]phenylmethylene]-2-oxo-,methyl ester, (3Z)-, ethanesulfonate (1:1)
Nintedanib esylate, 656247-18-6, UNII-42F62RTZ4G, , NSC753000, NSC-753000, KB-62821
Molecular Formula: C33H39N5O7S Molecular Weight: 649.75706
ニンテダニブエタンスルホン酸塩
Highly crystalline (mp = 305 °C) and exhibits a log P of 3.0 and good aqueous solubility (>20 mg/mL in water)…..J. Med. Chem., 2015, 58 (3), pp 1053–1063
Nintedanib esilate is a bright yellow powder soluble in water. The solubility increases at lower pH and decrease at higher pH due to the non-protonated free base which has a low solubility in water.At room temperature, the active substance exists only in one single crystalline form . The active substance contains no chiral centres. The double bond at C
-3 of the indole moiety allows forE/Zisomerism, but the activesubstance is the Z

Trade Name:Ofev® / Vargatef®
MOA:Tyrosine kinase inhibitor
Indication:Idiopathic pulmonary fibrosis (IPF); Non small cell lung cancer (NSCLC)
In 2011, orphan drug designation was assigned in the U.S. and Japan for the treatment of idiopathic pulmonary fibrosis. In 2013, orphan drug designation was also assigned for the same indication in the E.U. In 2014, a Breakthrough Therapy Designation was assigned to the compound for the treatment of idiopathic pulmonary fibrosis.
Nintedanib (formerly BIBF 1120) is a small molecule tyrosine-kinase inhibitor, targeting vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR) and platelet derived growth factor receptor (PDGFR) being developed by Boehringer Ingelheim as an anti-angiogenesis anti-cancer agent under the trade name Vargatef, and recently approved for treatment of idiopathic pulmonary fibrosis as Ofev.
The use of nintedanib or its salts, particularly its esylate salt is claimed for treating non-small cell lung cancer (NSCLC) in a patient who has received prior treatment with an anti-tumor therapy other than with nintedanib, wherein the patient to be treated is selected for treatment on the basis showing progression of the cancer within a period of 9 months or less after the initiation of said prior treatment. It is also claimed that the compound may be administered in combination with an anti-cancer drug, eg docetaxel. Nintedanib is known to be an antagonist of FGF-1, FGF-2, FGF-3, VEGF-1, VEGF-2, VEGF-3, PDGF-α and PDGF-β receptors.
Use of nintedanib for the treatment of non-small cell lung cancer in a patient who has received prior anti-tumour therapy other than with nintedanib. Boehringer Ingelheim has developed and launched Ofev, an oral capsule formulation of nintedanib, for the treatment of idiopathic pulmonary fibrosis (IPF), hepatic insufficiency and cancer, including metastatic NSCLC, ovarian, prostate and colorectal cancer. In October 2014, the US FDA approved the drug and an NDA was filed in Japan for IPF. Picks up from WO2014049099, claiming pharmaceutical combinations comprising nintedanib and sunitinib.
Nintedanib is an indolinone derivative angiogenesis inhibitor, originated at Boehringer Ingelheim. In 2014, the product candidate was approved and launched in the U.S. for the treatment of idiopathic pulmonary fibrosis, and a positive opinion was received by the EMA for the same indication. Also in 2014, Nintedanib was approved in the E.U. for the oral treatment of locally advanced, metastatic or locally recurrent non-small cell lung cancer (NSCLC) of adenocarcinoma tumour histology after first-line chemotherapy, in combination with docetaxel.The drug candidate is a small-molecule triple kinase inhibitor targeting the angiogenesis kinases (angiokinases) vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR) and platelet-derived growth factor receptor (PDGFR). By allowing the vascularization necessary for the nourishment of tumors, these angiokinases have been implicated in tumor growth, proliferation and metastasis. In previous studies, intedanib potently and selectively inhibited human endothelial cell proliferation and induced apoptosis in human umbilical vein endothelial cells (HUVEC). It showed good oral bioavailability and tolerance, and significant antitumor activity was observed in a number of human tumor xenograft models.
Mechanism of action
Nintedanib is an indolinone-derived drug that inhibits the process of blood vessel formation (angiogenesis). Angiogenesis inhibitors stop the formation and reshaping of blood vessels in and around tumours, which reduces the tumour’s blood supply, starving tumour cells of oxygen and nutrients leading to cell death and tumour shrinkage. Unlike conventional anti-cancer chemotherapy which has a direct cell killing effect on cancer cells, angiogenesis inhibitors starve the tumour cells of oxygen and nutrients which results in tumour cell death. One of the advantages of this method of anti-cancer therapy is that it is more specific than conventional chemotherapy agents, therefore results in fewer and less severe side effects than conventional chemotherapy.
The process of new blood vessel formation (angiogenesis) is essential for the growth and spread of cancers. It is mediated by signaling molecules (growth factors) released from cancer cells in response to low oxygen levels. The growth factors cause the cells of the tumour’s blood vessel to divide and reorganize resulting in the sprouting of new vessels in and around the tumour, improving its blood supply.
Angiogenesis is a process that is essential for the growth and spread of all solid tumours, blocking it prevents the tumour from growing and may result in tumour shrinkage as well as a reduction in the spread of the cancer to other parts of the body. Nintedanib exerts its anti-cancer effect by binding to and blocking the activation of cell receptors involved in blood vessel formation and reshaping (i.e. VEGFR 1-3, FGFR 1-3 AND PDGFRα and β). Inhibition of these receptors in the cells that make up blood vessels (endothelial cells, smooth muscle cells and pericytes) by Nintedanib leads to programmed cell death, destruction of tumor blood vessels and a reduction in blood flow to the tumour. Reduced tumour blood flow inhibits tumor cell proliferation and migration hence slowing the growth and spread of the cancer.[1]
Adverse effects
Preclinical studies have shown that nintedanib binds in a highly selective manner to the ATP binding pocked of its three target receptor families, without binding to similarly shaped ATP domains in other proteins, which reduces the potential for undesirable side effects.[2]
The most common side effects observed with nintedanib were reversible elevation in liver enzymes (10-28% of patients) and gastrointestinal disturbance (up to 50%). Side effects observed with nintedanib were worse with the higher 250 mg dose, for this reason subsequent trials have used the equally clinically effective 200 mg dose.[1][2][3][4][5][6][7][8][9]
Nintedanib inhibits the growth and reshaping of blood vessels which is also an essential process in normal wound healing and tissue repair. Therefore a theoretical side effect of nintedanib is reduced wound healing however, unlike other anti-angiogenic agents, this side effect has not been observed in patients receiving nintedanib.
Studies
Preclinical studies have demonstrated that nintedanib selectively binds to and blocks the VEGF, FGF and PDGF receptors, inhibiting the growth of cells that constitute the walls of blood vessels (endothelial and smooth muscle cells and pericytes) in vitro. Nintedanib reduces the number and density of blood vessels in tumours in vivo, resulting in tumour shrinkage.[1][2] Nintedanib also inhibits the growth of cells that are resistant to existing chemotherapy agents in vitro, which suggests a potential role for the agent in patients with solid tumours that are unresponsive to or relapse following current first line therapy.[10]
Early clinical trials of nintedanib have been carried out in patients with non-small cell lung, colorectal, uterine, endometrial, ovarian and cervical cancer and multiple myeloma.[4][5][7][8][9] These studies reported that the drug is active in patients, safe to administer and is stable in the bloodstream. They identified that the maximum tolerated dose of nintedanib is 20 0 mg when taken once a day.
Clinical studies
In the first human trials, nintedanib halted the growth of tumours in up to 50% of patients with non-small cell lung cancer and 76% of patients with advanced colorectal cancer and other solid tumours.[4][8] A complete response was observed in 1/26 patients with non-small cell lung and 1/7 patients with ovarian cancer treated with nintedanib. A further 2 patients with ovarian cancer had partial responses to nintedanib.[8][9]
Two phase II trials have been carried out assessing the efficacy, dosing and side effects of nintedanib in non-small cell lung and ovarian cancer. These trials found that nintedanib delayed relapse in patients with ovarian cancer by two months[6] and that overall survival of patients with non-small cell lung who received nintedanib was similar to that observed with the FDA approved VEGFR inhibitor sorafenib. These trials also concluded that increasing the dose of the nintedanib has no effect on survival.[3]
SYNTHESIS

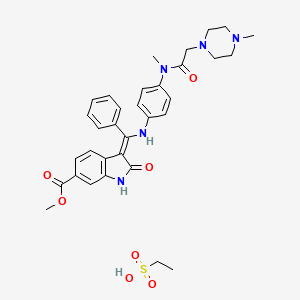
WO2009071523A1
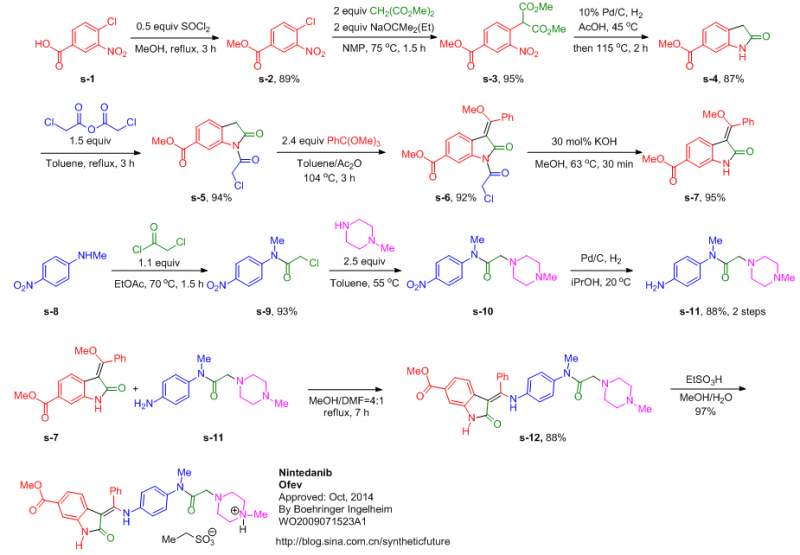
MORE SYNTHESIS
Route 1

Reference:1. WO0127081A1.
2. US6762180B1.
3. J. Med. Chem. 2009, 52, 4466-4480.
Route 2

Reference:1. WO2009071523A1 / US8304541B2.
Route 3

Reference:1. CN104262232A.
Route 4

Reference:1. CN104844499A.
Current clinical trials
Nintedanib is being tested in several phase I to III clinical trials for cancer. Angiogenesis inhibitors such as nintedanib may be effective in a range of solid tumour types including; lung, ovarian, metastatic bowel, liver and brain cancer. Patients are also being recruited for three phase III clinical trials that will evaluate the potential benefit of nintedanib when added to existing 1st line treatments in patients with ovarian.[11] and 2nd line treatment in non-small cell lung cancer [12][13] The phase III trials of nintedanib in lung cancer have been named LUME-Lung 1 and LUME-Lung 2.
Current phase II trials are investigating the effect of nintedanib in patients with metastatic bowel cancer, liver cancer and the brain tumour: glioblastoma multiforme.[14]
Phase III trials are investigating the use of nintedanib in combination with the existing chemotherapy agents permexetred and docetaxel in patients with non-small cell lung cancer,[15] and in combination with carboplatin and paclitaxel as a first line treatment for patients with ovarian cancer.[16]
A phase III clinical trial was underway examining the safety and efficacy of nintedanib on patients with the non-cancerous lung condition idiopathic pulmonary fibrosis.[17] Nintedanib, under the brand name Ofev, was approved by the FDA for treatment of idiopathic pulmonary fibrosis on 15 Oct 2014. [18]
In terms of clinical development, additional phase III clinical trials are ongoing for the treatment of epithelial ovarian cancer, fallopian tube or primary peritoneal cancer, in combination with chemotherapy, and for the treatment of refractory metastatic colorectal cancer. Phase II clinical trials are also ongoing at the company for the treatment of glioblastoma multiforme, previously untreated patients with renal cell cancer, and for the treatment of patients with unresectable malignant pleural mesothelioma. The National Cancer Center of Korea (NCC) is evaluating the compound in phase II studies as second line treatment for small cell lung cancer (SCLC). The Centre Oscar Lambret is also conducting phase II clinical trials for the treatment of breast cancer in combination with docetaxel. Phase II trials are under way at EORTC as second line therapy for patients with either differentiated or medullary thyroid cancer progressing after first line therapy. The compound is also in early clinical development for the treatment of cancer of the peritoneal cavity, hepatocellular carcinoma, acute myeloid leukemia and ovarian cancer. Clinical trials have been completed for the treatment of prostate cancer and for the treatment of colorectal cancer. Boehringer Ingelheim is also conducting phase I/II clinical trials for the treatment of NSCLC and acute myeloid leukemia in addition to low-dose cytarabine. Phase I clinical studies are ongoing at the company for the treatment of epithelial ovary cancer and for the treatment of patients with mild and moderate hepatic impairment. The company had been evaluating the compound in early clinical trials for the treatment of prostate cancer in combination with docetaxel, but recent progress reports for this indication are not available at present.
In 2011, orphan drug designation was assigned in the U.S. and Japan for the treatment of idiopathic pulmonary fibrosis. In 2013, orphan drug designation was also assigned for the same indication in the E.U. In 2014, a Breakthrough Therapy Designation was assigned to the compound for the treatment of idiopathic pulmonary fibrosis.
PAPER
http://pubs.acs.org/doi/full/10.1021/jm501562a
Nintedanib: From Discovery to the Clinic
†Department of Medicinal Chemistry; §Department of Drug Metabolism and Pharmacokinetics; ‡Department of Non-Clinical Drug Safety; ∥Department of Translational Medicine and Clinical Pharmacology; ⊥Department of Respiratory Diseases Research; and #Corporate Division Medicine, TA Oncology, Boehringer Ingelheim Pharma GmbH & Co. KG, 88397 Biberach an der Riss, Germany
○ Clinical Development and Medical Affairs, Respiratory, Boehringer Ingelheim Inc., Ridgefield, Connecticut 06877, United States
∇ Boehringer Ingelheim RCV GmbH & Co. KG, A-1121 Vienna, Austria
J. Med. Chem., 2015, 58 (3), pp 1053–1063
DOI: 10.1021/jm501562a
Nintedanib (BIBF1120) is a potent, oral, small-molecule tyrosine kinase inhibitor, also known as a triple angiokinase inhibitor, inhibiting three major signaling pathways involved in angiogenesis. Nintedanib targets proangiogenic and pro-fibrotic pathways mediated by the VEGFR family, the fibroblast growth factor receptor (FGFR) family, the platelet-derived growth factor receptor (PDGFR) family, as well as Src and Flt-3 kinases. The compound was identified during a lead optimization program for small-molecule inhibitors of angiogenesis and has since undergone extensive clinical investigation for the treatment of various solid tumors, and in patients with the debilitating lung disease idiopathic pulmonary fibrosis (IPF). Recent clinical evidence from phase III studies has shown that nintedanib has significant efficacy in the treatment of NSCLC, ovarian cancer, and IPF. This review article provides a comprehensive summary of the preclinical and clinical research and development of nintedanib from the initial drug discovery process to the latest available clinical trial data.
-
Roth, G. J.; Heckel, A.; Colbatzky, F.; Handschuh, S.; Kley, J.; Lehmann-Lintz, T.; Lotz, R.; Tontsch-Grunt,U.; Walter, R.; Hilberg, F.Design, synthesis, and evaluation of indolinones as triple angiokinase inhibitors and the discovery of a highly specific 6-methoxycarbonyl-substituted indolinone (BIBF 1120) J. Med. Chem.2009, 52, 4466– 4480
-
2.Roth, G. J.; Sieger, P.; Linz, G.; Rall, W.; Hilberg, F.; Bock, T. 3-Z-[1-(4-(N-((4-Methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone monoethanesulphonate and the use thereof as a pharmaceutical composition. WO2004/013099. 2004.
-
3.Merten, J.; Linz, G.; Schnaubelt, J.; Schmid, R.; Rall, W.; Renner, S.; Reichel, C.; Schiffers, R. Process for the manufacture of an indolinone derivative. WO2009/071523.
2009
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm900431g
J. Med. Chem., 2009, 52 (14), pp 4466–4480
DOI: 10.1021/jm900431g

Inhibition of tumor angiogenesis through blockade of the vascular endothelial growth factor (VEGF) signaling pathway is a new treatment modality in oncology. Preclinical findings suggest that blockade of additional pro-angiogenic kinases, such as fibroblast and platelet-derived growth factor receptors (FGFR and PDGFR), may improve the efficacy of pharmacological cancer treatment. Indolinones substituted in position 6 were identified as selective inhibitors of VEGF-, PDGF-, and FGF-receptor kinases. In particular, 6-methoxycarbonyl-substituted indolinones showed a highly favorable selectivity profile. Optimization identified potent inhibitors of VEGF-related endothelial cell proliferation with additional efficacy on pericyctes and smooth muscle cells. In contrast, no direct inhibition of tumor cell proliferation was observed. Compounds 2 (BIBF 1000) and 3 (BIBF 1120) are orally available and display encouraging efficacy in in vivo tumor models while being well tolerated. The triple angiokinase inhibitor 3 is currently in phase III clinical trials for the treatment of nonsmall cell lung cancer.
PATENT
WO-2014180955
The present invention relates to a beneficial treatment of tumours in patients suffering from NSCLC, and to a clinical marker useful as predictive variable of the responsiveness of tumours in patients suffering from NSCLC. The present invention further relates to a method for selecting patients likely to respond to a given therapy, wherein said method optionally comprises the use of a specific clinical marker. The present invention further relates to a method for delaying disease progression and/or prolonging patient survival of NSCLC patients, wherein said method comprises the use of a specific clinical marker.
The monoethanesulphonate salt form of this compound presents properties which makes this salt form especially suitable for development as medicament. The chemical structure of 3-Z-[l-(4-(N-((4-methyl-piperazin-l-yl)-methylcarbonyl)-N-methyl-amino)-anilino)- 1 -phenyl-methylene] -6-methoxycarbonyl-2-indolinone-monoethanesulphonate (ΓΝΝ name nintedanib esylate) is depicted below as Formula Al .
Formula Al

This compound is thus for example suitable for the treatment of diseases in which angiogenesis or the proliferation of cells is involved. The use of this compound for the treatment of immunologic diseases or pathological conditions involving an
immunologic component is being described in WO 2004/017948, the use for the treatment of, amongst others, oncological diseases, alone or in combination, is being described in WO 2004/096224 and WO 2009/147218, and the use for the treatment of fibrotic diseases is being described in WO 2006/067165.
A method using biomarkers for monitoring the treatment of an individual with the compound 3-Z-[l-(4-(N-((4-methyl-piperazin-l-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-l -phenyl-methylene] -6-methoxycarbonyl-2-indolinone or a pharmaceutically acceptable salt thereof, wherein it is determined if a sample from said individual comprises a biomarker in an amount that is indicative for said treatment, is disclosed in WO 2010/103058.
PATENT
http://www.google.com/patents/US20110201812
The present invention relates to a process for the manufacture of a specific indolinone derivative and a pharmaceutically acceptable salt thereof, namely 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone and its monoethanesulfonate, to new manufacturing steps and to new intermediates of this process.
The indolinone derivative 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone and its monoethanesulfonate are known from the following patent applications: WO 01/027081, WO 04/013099, WO 04/017948, WO 04/096224 and WO 06/067165. These patent applications disclose the compound, a process for its manufacture, a specific salt form of this compound and the use of the compound or its salt in a pharmaceutical composition to treat oncological or non-oncological diseases via inhibition of the proliferation of target cells, alone or in combination with further therapeutic agents. The mechanism of action by which the proliferation of the target cells occurs is essentially a mechanism of inhibition of several tyrosine kinase receptors, and especially an inhibition of the vascular endothelial growth factor receptor (VEGFR).




EXAMPLE 1Synthesis of the 6-methoxycarbonyl-2-oxindole in accordance with the process shown in synthesis scheme CSynthesis of benzoic acid, 4-chloro-3-nitro-, methylester
-
- 20 kg of 4-chloro-3-nitro-benzoic acid (99.22 mol) is suspended in 76 L methanol. 5.9 kg thionylchloride (49.62 mol) is added within 15 minutes and refluxed for about 3 hours. After cooling to about 5° C., the product is isolated by centrifugation and drying at 45° C.
- Yield: 19.0 kg (88.8% of theoretical amount)
- Purity (HPLC): 99.8%
Synthesis of propanedioic acid, [4-(methoxycarbonyl)-2-nitrophenyl]-, dimethylester
-
- 12.87 kg of malonic acid, dimethylester (97.41 mol) is added to a hot solution (75° C.) of 10.73 kg sodium-tert.amylate (97.41 mol) in 35 L 1-methyl-2-pyrrolidinone (NMP). A solution of 10 kg benzoic acid, 4-chloro-3-nitro-, methylester (46.38 mol) in 25 L 1-methyl-2-pyrrolidinone is added at 75° C. After stirring for 1.5 hours at about 75° C. and cooling to 20° C., the mixture is acidified with 100 L diluted hydrochloric acid to pH 1. After stirring for 1.5 hours at about 5° C., the product is isolated by centrifugation and drying at 40° C.
- Yield: 13.78 kg (95.4% of theoretical amount)
- Purity (HPLC): 99.9%
- Alternatively, propanedioic acid, [4-(methoxycarbonyl)-2-nitrophenyl]-, dimethylester can be synthesized as follows:
- 33.1 kg of malonic acid, dimethylester (250.6 mol) and 27.0 kg benzoic acid, 4-chloro-3-nitro-, methylester (125.3 mol) are subsequently added to a solution of 45.1 kg sodium-methylate (250.6 mol) in 172 kg 1-methyl-2-pyrrolidinone (NMP) at 20° C. After stirring for 1.5 hours at about 45° C. and cooling to 30° C., the mixture is acidified with 249 L diluted hydrochloric acid. At the same temperature, the mixture is seeded, then cooled to 0° C. and stirred for an additional hour. The resulting crystals are isolated by centrifugation, washed and dryed at 40° C.
- Yield: 37.5 kg (86% of theoretical amount)
- Purity (HPLC): 99.7%
Synthesis of 6-methoxycarbonyl-2-oxindole
A solution of 13 kg propanedioic acid, [4-(methoxycarbonyl)-2-nitrophenyl]-, dimethylester (41.77 mol) in 88 L acetic acid is hydrogenated at 45° C. and under 40-50 psi in the presence of 1.3 kg Pd/C 10%. After standstill of the hydrogenation, the reaction is heated up to 115° C. for 2 hours. The catalyst is filtered off and 180 L water is added at about 50° C. The product is isolated after cooling to 5° C., centrifugation and drying at 50° C.
-
- Yield: 6.96 kg (87.2% of theoretical amount)
- Purity (HPLC): 99.8%
EXAMPLE 2Synthesis of the “chlorimide” (methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate)
Method 1
6-methoxycarbonyl-2-oxindole (400 g; 2.071 mol) is suspended in toluene (1200 ml) at room temperature. Chloroacetic anhydride (540 g; 3.095 mol) is added to this suspension. The mixture is heated to reflux for 3 h, then cooled to 80° C. and methyl cyclohexane (600 ml) is added within 30 min. The resulting suspension is further cooled down to room temperature within 60 min. The mother liquor is separated and the solid is washed with ice cold methanol (400 ml). The crystals are dried to afford 515.5 g (93.5%) of the “chlorimide” compound as a white solid. 1H-NMR (500 MHz, DMSO-d6) δ: 8.66 (s, 1H, 6-H); 7.86 (d, J=8.3 Hz, 1H, 8-H); 7.52 (d, J=8.3 Hz, 1H, 9-H); 4.98 (s, 2H, 15-H2); 3.95 (s, 3H, 18-H3); 3.88 (s, 2H, 3-H2). 13C-NMR (126 MHz, DMSO-d6) δ: 174.7 (C-2); 36.0 (C-3); 131.0 (C-4); 140.8 (C-5); 115.7 (C-6); 128.9 (C-7); 126.1 (C-8); 124.6 (C-9); 166.6 (C-10); 165.8 (C-13); 46.1 (C-15); 52.3 (C-18). MS: m/z 268 (M+H)+. Anal. calcd. for C12H10ClNO4: C, 53.85; H, 3.77; Cl, 13.25; N, 5.23. Found: C, 52.18; H, 3.64; Cl, 12.89; N, 5.00.
Method 2
6-Methoxycarbonyl-2-oxindole (10 g; 0.052 mol) is suspended in n-butyl acetate (25 ml) at room temperature. To this suspension a solution of chloroacetic anhydride (12.8 g; 0.037 mol) in n-butyl acetate (25 ml) is added within 3 min. The mixture is heated to reflux for 2 h, then cooled to 85° C. and methyl cyclohexane (20 ml) is added. The resulting suspension is further cooled down to room temperature and stirred for 2 h. The mother liquor is separated and the solid is washed with methanol (400 ml) at ambient temperature. The crystals are dried to afford 12.7 g (91.5%) of the “chlorimide” compound as a slightly yellow solid.
EXAMPLE 3Synthesis of the “chlorenol” (methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate)
Method 1
Methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate (12.0 g; 0.045 mol) is suspended in toluene (60 ml) at ambient temperature. Acetic anhydride (16.2 g; 0.157 mol) is added to this suspension. The mixture is heated to not less than 104° C. and trimethyl orthobenzoate (20.0 g; 0.108 mol) is added within 60 min. During the addition period and subsequent stirring at the same temperature for 3 h, volatile parts of the reaction mixture are distilled off. The concentration of the reaction mixture is kept constant by replacement of the distilled part by toluene (40 ml). The mixture is cooled down to 5° C., stirred for 1 h and filtrated. The solid is subsequently washed with toluene (14 ml) and with a mixture of toluene (8 ml) and ethyl acetate (8 ml). After drying, 16.3 g (91.7%) of the “chlorenol” compound are isolated as slightly yellow crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 8.73 (d, J=1.5 Hz, 1H, 6-H); 8.09 (d, J=8.0 Hz, 1H, 9-H); 7.90 (dd, J=8.1; 1.5 Hz, 1H, 8-H); 7.61-7.48 (m, 5H, 21-H, 22-H, 23-H, 24-H, 25-H); 4.85 (s, 2H, 18-H2); 3.89 (s, 3H, 27-H3); 3.78 (s, 3H, 15-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 165.9 (C-2+C16); 103.9 (C-3); 127.4; 128.6; 130.0; 135.4 (C-4+C-5+C-7+C-20); 115.1 (C-6); 126.1 (C-8); 122.5 (C-9); 166.7 (C-10); 173.4 (C-13); 58.4 (C-15); 46.4 (C-18); 128.6 (C-21+C-22+C-24+C-25); 130.5 (C-23); 52.2 (C-27). MS: m/z 386 (M+H)+. Anal. calcd. for C20H16ClNO5: C, 62.27; H, 4.18; Cl, 9.19; N, 3.63. Found: C, 62.21; H, 4.03; Cl, 8.99; N, 3.52.
Method 2
Methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate (12.0 g; 0.045 mol) is suspended in xylene (60 ml) at ambient temperature. Acetic anhydride (16.2 g; 0.157 mol) is added to this suspension. The mixture is heated to reflux, trimethyl orthobenzoate (20.0 g; 0.108 mol) is added within 40 min and heating is maintained for 4 h. The mixture is cooled down to 0° C. and the mother liquor is separated. The solid is subsequently washed with xylene (14 ml) and a mixture of xylene (8 ml) and ethyl acetate (8 ml). After drying 14.3 g (81.0%) of the “chlorenol” compound are isolated as yellow crystals.
Method 3
Methyl-1-(chloroacetyl)-2-oxoindoline-6-carboxylate (12.0 g; 0.045 mol) is suspended in toluene (60 ml) at ambient temperature. Acetic anhydride (16.2 g; 0.157 mol) is added to this suspension. The mixture is heated to reflux, trimethyl orthobenzoate (20.0 g; 0.108 mol) is added within 40 min and heating is maintained for 3 h. The mixture is cooled down to 0° C. and the mother liquor is separated. The solid is subsequently washed with toluene (14 ml) and a mixture of toluene (8 ml) and ethyl acetate (8 ml). After drying 15.3 g (87.3%) of the “chlorenol” compound are isolated as fawn crystals.
EXAMPLE 4Synthesis of the “enolindole” (methyl-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate)
Method 1
A solution of potassium hydroxide (0.41 g, 0.006 mol) in methanol (4 ml) is added at 63° C. to a suspension of methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (8.0 g; 0.020 mol) in methanol (32 ml). The mixture is then stirred for 30 min, cooled to 0° C. and stirring is maintained for 2 h. After filtration, the solid is washed with methanol (24 ml) and dried to afford 6.0 g (94.6%) of the “enolindole” compound as yellow crystals. 1H-NMR (500 MHz, CDCl3) δ: 8.08 (s, 1H, 1-H); 7.88 (d, J=7.8 Hz, 1H, 9-H); 7.75 (m, 1H, 8-H); 7.52-7.56 (m, 3H, 18-H, 19-H, 20-H); 7.40-7.45 (m, 3H, 6-H, 17-H, 21-H); 3.92 (s, 3H, 23-H3); 3.74 (s, 3H, 13-H3). 13C-NMR (126 MHz, CDCl3) δ: 168.8 (C-2); 107.4 (C-3); 130.8 (C-4); 138.2 (C-5); 109.4 (C-6); 128.2 and 128.3 (C-7, C-16); 123.5 (C-8); 123.1 (C-9); 170.1 (C-11); 57.6 (C-13); 167.2 (C-14); 128.7 and 128.9 (C-17, C-18, C-20, C-21); 130.5 (C-19); 52.1 (C-23). MS (m/z): 310 (M+H)+. Anal. calcd. for C18H15NO4: C, 69.89; H, 4.89; N, 4.53. Found: C, 69.34; H, 4.92; N, 4.56.
Method 2
A suspension of methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (7.0 g; 0.018 mol) in methanol (28 ml) is heated to reflux. Within 3 min, a solution of sodium methoxide in methanol (0.24 g, 30 (w/w), 0.001 mol) is added to this suspension. The mixture is then stirred for 30 min, cooled to 5° C. and stirring is maintained for 2 h. After filtration, the solid is washed with methanol (9 ml) and dried to afford 5.4 g (89.7%) of the “enolindole” compound as yellow crystals.
Method 3
A suspension of methyl-1-(chloroacetyl)-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (8.0 g; 0.021 mol) in methanol (32 ml) is heated to reflux. A solution of sodium methoxide in methanol (0.74 g, 30% (w/w), 0.004 mol), further diluted with methanol (4 ml), is added dropwise to this suspension. The mixture is then stirred for 90 min, cooled to 0° C. and stirring is maintained for 2 h. After filtration, the solid is washed with methanol (24 ml) and dried to afford 5.9 g (91.2%) of the “enolindole” compound as yellow crystals.
EXAMPLE 5Synthesis of the “chloroacetyl” (N-(4-nitroanilino)-N-methyl-2-chloro-acetamide)
Method 1
A suspension of N-methyl-4-nitroaniline (140 g; 0.920 mol) in ethyl acetate (400 ml) is heated to 70° C. Within 90 min, chloro acetylchloride (114 g; 1.009 mol) is added to this suspension. The resulting solution is then refluxed for 1 h, cooled to 60° C. and methyl cyclohexane (245 ml) is added. The suspension is further cooled down to 0° C. and stirred for 1 h. The reaction mixture is filtrated, washed with methyl cyclohexane (285 ml) and the precipitate is dried to afford 210.4 g (92.7%) of the “chloroacetyl” compound as white crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 8.29 (d, J=8.5 Hz, 2H, 1-H+3-H); 7.69 (d, J=8.5 Hz, 2H, 4-H+6-H); 4.35 (s, 2H, 9-H2); 3.33 (s, 3H, 12-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 124.6 (C-1+C-3); 145.6 (C-2); 127.4 (C-4+C-6); 148.6 (C-5); 165.6 (C-8); 42.7 (C-9); 37.2 (C-12). MS (m/z): 229 (M+H)+. Anal. calcd. for C9H9ClN2O3: C, 47.28; H, 3.97; N, 12.25. Found: C, 47.26; H, 3.99; Cl, 15.73; N, 12.29.
Method 2
A suspension of N-methyl-4-nitroaniline (20.0 g; 0.131 mol) in ethyl acetate (20 ml) is heated to 60° C. Within 15 min, a solution of chloro acetic anhydride (26.0 g; 0.151 mol) in ethyl acetate (60 ml) is added to this suspension. The resulting solution is then refluxed for 1 h, cooled to 75° C. ° C. and methyl cyclohexane (80 ml) is added. After seeding at 60° C., the suspension is further cooled down to 0° C. and stirred for 1 h. The reaction mixture is filtrated, washed with methyl cyclohexane (40 ml) and the precipitate is dried to afford 25.9 g (83.3%) of the “chloroacetyl” compound as grey crystals.
EXAMPLE 6Synthesis of the “nitroaniline” (N-(4-nitrophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide) and of the “aniline” (N-(4-aminophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide)
Method 1
A suspension of N-(4-nitroanilino)-N-methyl-2-chloro-acetamide (20.0 g; 0.087 mol) in toluene (110 ml) is heated to 40° C. Within 30 min, 1-methylpiperazine (21.9 g; 0.216 mol) is added dropwise. After purging of the dropping funnel with toluene (5 ml) the reaction mixture is stirred for 2 h at 55° C., cooled to ambient temperature and washed with water (15 ml). The organic layer is diluted with isopropanol (100 ml) and Pd/C (10%; 1.0 g) is added. After subsequent hydrogenation (H2, 4 bar) at 20° C. the catalyst is removed. Approximately ⅘ of the volume of the resulting solution is evaporated at 50° C. The remaining residue is dissolved in ethyl acetate (20 ml) and toluene (147 ml) heated to 80° C., then cooled to 55° C. and seeded. The reaction mixture is further cooled to 0° C. and stirred for 3 h at the same temperature. After filtration, the solid is washed with ice cold toluene (40 ml) and dried to afford 20.2 g (88.0%) of the “aniline” compound as white crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 6.90 (d, J=8.5 Hz, 2H, 4-H+6-H); 6.65 (d, J=8.5 Hz, 2H, 1-H+3-H); 5.22 (2H, 19-H2); 3.04 (s, 3H, 9-H3); 2.79 (s, 2H, 11-H2); 2.32 (m, 4H, 13-H2+17-H2); 2.23 (m, 4H, 14-H2+16-H2); 2.10 (s, 3H, 18-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 114.0 (C-1+C-3); 148.0 (C-2); 127.6 (C-4+C-6); 131.5 (C-5); 168.9 (C-8); 36.9 (C-9); 58.5 (C-11); 52.4 (C-13+C-17); 54.6 (C-14+C-16); 45.7 (C-18). MS (m/z): 263 (M+H)+. Anal. calcd. for C14H22N4O: C, 64.09; H, 8.45; N, 21.36. Found: C, 64.05; H, 8.43; N, 21.39.
Method 2
A suspension of N-(4-nitroanilino)-N-methyl-2-chloro-acetamide (14.5 g; 0.063 mol) in ethyl acetate (65 ml) is heated to 40° C. Within 30 min, 1-methylpiperazine (15.8 g; 0.156 mol) is added dropwise. After purging of the dropping funnel with ethyl acetate (7 ml) the reaction mixture is stirred at 50° C. for 90 min, cooled to ambient temperature and washed with water (7 ml). The organic layer is diluted with isopropanol (75 ml) and dried over sodium sulphate. After separation of the solid, Pd/C (10%; 2.0 g) is added and the solution is hydrogenated (H2, 5 bar) at ambient temperature without cooling. Subsequently the catalyst is removed by filtration and the solvent is evaporated at 60° C. The remaining residue is dissolved in ethyl acetate (250 ml) and recrystallized. After filtration and drying 10.4 g (60.4%) of the “aniline” compound are isolated as white crystals.
EXAMPLE 7Synthesis of the “anilino” (3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone)
Method 1
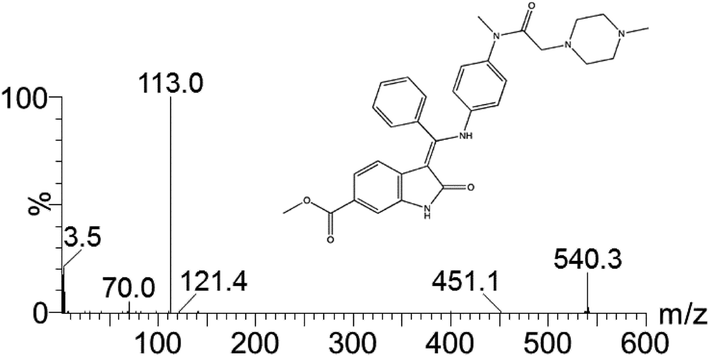
A suspension of methyl-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (10.0 g; 0.032 mol) and N-(4-aminophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide (8.6 g; 0.032 mol) in a mixture of methanol (72 ml) and N,N-dimethylformamide (18 ml) is heated to reflux. After 7 h of refluxing the suspension is cooled down to 0° C. and stirring is maintained for additional 2 h. The solid is filtered, washed with methanol (40 ml) and dried to afford 15.4 g (88.1%) of the “anilino” compound as yellow crystals. 1H-NMR (500 MHz, DMSO-d6) δ: 11.00 (s, 1H, 23-H); 12.23 (s, 19-H); 7.61 (t; J=7.1 Hz, 1H, 33-H); 7.57 (t, J=7.5 Hz, 2H, 32-H+34-H); 7.50 (d, J=7.7 Hz, 2H, 31-H+35-H); 7.43 (d, J=1.6 Hz, 1H, 29-H); 7.20 (dd, J=8.3; 1.6 Hz, 1H, 27-H); 7.13 (d, J=8.3 Hz, 2H, 14-H+18-H); 6.89 (d, 8.3 Hz, 2H, 15-H+17-H); 5.84 (d, J=8.3 Hz, 1H, 26-H); 3.77 (s, 3H, 40-H3); 3.06 (m, 3H, 12-H3); 2.70 (m, 2 H, 8-H2); 2.19 (m, 8H, 2-H2, 3-H2, 5-H2, 6-H2); 2.11 (s, 3H, 7-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 54.5 (C-2+C-6); 52.2 (C-3+C-5); 45.6 (C-7); 59.1 (C-8); 168.5 (C-9); 36.6 (C-12); 140.1 (C-13); 127.6 (C-14+C-18); 123.8 (C-17+C-15); 137.0 (C-16); 158.3 (C-20); 97.5 (C-21); 170.1 (C-22); 136.2 (C-24); 128.9 (C-25); 117.2 (C-26); 121.4 (C-27); 124.0 (C-28); 109.4 (C-29); 131.9 (C-30); 128.4 (C-31+C-35); 129.4 (C-32+C-34); 130.4 (C-33); 166.3 (C-37); 51.7 (C-40). MS (m/z): 540 (M+H)+. Anal. calcd. for C31H33N5O4: C, 69.00; H, 6.16; N, 12.98. Found: C, 68.05; H, 6.21; N, 12.81.
Method 2
A suspension of methyl-3-[methoxy(phenyl)methylene]-2-oxoindoline-6-carboxylate (20.0 g; 0.064 mol) and N-(4-aminophenyl)-N-methyl-2-(4-methylpiperazin-1-yl)acetamide (17.1 g; 0.065 mol) in methanol (180 ml) is heated to reflux for 7.5 h. The resulting suspension is cooled down to 10° C. within 1 h and stirring is maintained for 1 h. After filtration, the solid is washed with ice cold methanol (80 ml) and dried to afford 31.0 g (89.0%) of the “anilino” compound as yellow crystals.
EXAMPLE 8Synthesis of the 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone, monoethanesulfonate

A suspension of 3-Z-[1-(4-(N-((4-methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone (30.0 g; 0.055 mol) in methanol (200 ml) and water (2.4 ml) is heated to 60° C. Aqueous ethanesulfonic acid (70% (w/w); 8.75 g; 0.056 mol) is added to the reaction mixture. The resulting solution is cooled to 50° C., seeded and then diluted with isopropanol (200 ml). The mixture is further cooled to 0° C. and stirred for 2 h at this temperature. The precipitate is isolated, washed with isopropanol (120 ml) and dried to furnish 35.1 g (97.3%) of the monoethanesulfonate salt of the compound as yellow crystals. 1H-NMR (400 MHz, DMSO-d6) δ: 12.26 (s, 11-H); 10.79 (s, 1H, 1-H); 9.44 (s, 1H, 24-H); 7.64 (m, 1H, 32-H); 7.59 (m, 2H, 31-H+33-H); 7.52 (m, 2H, 30-H+34-H); 7.45 (d, J=1.6 Hz, 1H, 7-H); 7.20 (dd, J=8.2; 1.6 Hz, 1H, 5-H); 7.16 (m, 2H, 14-H+16-H); 6.90 (m, 2H, 13-H+17-H); 5.85 (d, J=8.2 Hz, 1H, 4-H); 3.78 (s, 3H, 37-H3); 3.45-2.80 (broad m, 4H, 23-H2+25-H2); 3.08 (s, 3H, 28-H3); 2.88 (s, 2H, 20-H2); 2.85-2.30 (broad m, 4H, 22-H2+26-H2); 2.75 (s, 3H, 27-H3); 2.44 (q, J=7.4 Hz, 2H, 39-H2); 1.09 (t, J=7.4 Hz, 3H, 38-H3). 13C-NMR (126 MHz, DMSO-d6) δ: 9.8 (C-38); 36.6 (C-28); 42.3 (C-27); 45.1 (C-39); 51.7 (C-37); 48.9 (C-22+C-26); 52.6 (C-23+C-25); 57.5 (C-20); 97.7 (C-3); 109.5 (C-7); 117.3 (C-4); 121.4 (C-5); 123.8 (C-13+C-17); 124.1 (C-6); 127.7 (C-14+C-16); 128.4 (C-30+C-34); 128.8 (C-9); 129.5 (C-31+C-33); 130.5 (C-32); 132.0 (C-29); 168.5 (C-9); 136.3 (C-8); 137.3 (C-12); 139.5 (C-15); 158.1 (C-10); 166.3 (C-35); 168.0 (C-19); 170.1 (C-2). MS (m/z): 540 (M(base)+H)+. Anal. calcd. for C33H39N5O7S: C, 60.17; H, 6.12; N, 10.63; S, 4.87. Found: C, 60.40; H, 6.15; N, 10.70; S, 4.84.
CLIPS

After a classical malonic ester addition to arene 3, the resulting nitro benzene (4) is hydrogenated under acidic conditions, furnishing the 6-methoxycarbonyl-substituted oxindole 5 via decarboxylative cyclization. Condensation of 5 with trimethyl orthobenzoate in acetic anhydride leads to compound 6, one of the two key building blocks of the synthesis. The concomitant N-acetylation of the oxindole activates the scaffold for the condensation reaction.
The aniline side chain (9) can be prepared by a one-pot bromo-acetylation/amination of the para-nitro-phenylamine (7) using bromoacetyl bromide and N-methylpiperazine and a subsequent hydrogenation furnishing 9 as the second key building block. Condensation of both building blocks in an addition–elimination sequence and subsequent acetyl removal with piperidine furnishes 2 as free base (pKa = 7.9), which subsequently is converted into its monoethanesulfonate salt (1). Compound 1 is highly crystalline (mp = 305 °C) and exhibits a log P of 3.0 and good aqueous solubility (>20 mg/mL in water).
CLIPS
see
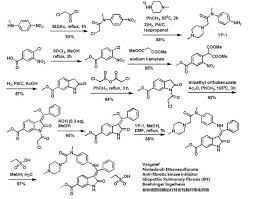
CLICK ON PIC
Updates………..
“J.Med.Chem” 2009 Vol. 52, page 4466-4480 and the “Chinese Journal of Pharmaceuticals” 2012, Vol. 43, No. 9, page 726-729 reported a further intermediate A and B synthesis, and optimized from the reaction conditions, the reaction sequence, the feed ratio and catalyst selection, etc., so that the above-described synthetic routes can be simplified and reasonable.
PATENT
CN105461609A
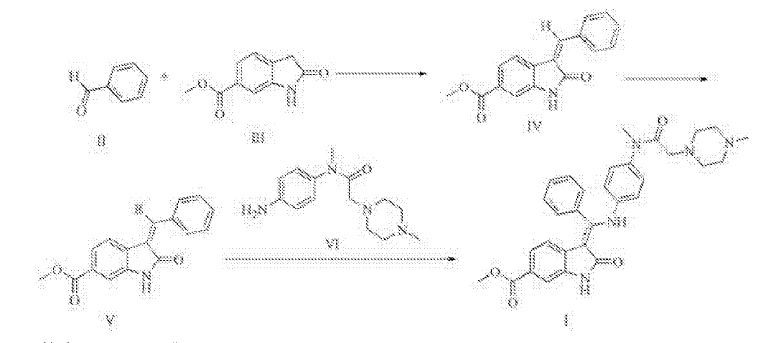
MACHINE TRANSLATED FROM CHINESE
Synthesis of Trinidad Neeb (I),
A 500ml reaction flask was charged 30g of compound V, 22.5g compound of the VI, ethanol 300ml, sodium bicarbonate and 15g, the reaction was heated to reflux for 2 hours, the reaction mixture was added to 600ml of water, there are large amount of solid precipitated, was filtered, the cake washed with 100ml washed once with methanol, a yellow solid 41.9g refined Trinidad Neeb (I). Yield 92.7%.
4 bandit R (400MHz, dmso) δ11 · 97 (s, 1H), 8.38 (s, 1H), 7.97 (dd, J = 11.9, 5.0Hz, 2H), 7.67 (d, J = 8.1Hz, 1H), 7.16 (ddd, J = 26.9, 22.1, 7.0Hz, 5H), 6.85 (d, J = 8.6Hz, 2H), 6.63 (d, J = 8.7Hz, 2H), 3.90 (s, 3H), 2.99 (s, 3H), 2.69 (s, 2H), 2.51-2.24 (m, 8H), 2.20 (s, 3H) MS:. m / z540 (m + 1) + 2 Example: Preparation of compound IV 250ml reaction flask was added 28.7g of 2- oxindole-6-carboxylate, 130ml ethanol, stirred open, then added 30.3ml (31.8g) benzaldehyde, 2.97 mL piperidine was heated to 70 ° C-80 after ° C for 2 hours, allowed to cool to 20 ° C- 30 ° C, the precipitate was filtered, the filter cake was washed with absolute ethanol, 50 ° C 5 hours and dried in vacuo give a yellow solid 38.7g (IV of), yield: 92.4% Preparation of compound V square in 500ml reaction flask was added 30g compound IV, dichloromethane 360ml, cooled with ice water to 0-5 ° C, 71/92 bromine 3.lml (9.7g), drop finished warmed to 20- 30 ° C, 3 hours after the reaction, the reaction solution was washed once with 150ml dichloromethane layer was concentrated oil was done by adding 200ml ethanol crystallization, filtration, 60 ° C and dried under vacuum 36.lg white solid (V ), yield: 93 · 8%.
After Trinidad Technip (I) are synthesized in the reaction flask was added 500ml of 30g compound V, 33.0g compound of the VI, ethanol 300ml, sodium bicarbonate, 15g, was heated to reflux for 2 hours, the reaction mixture was added to 600ml water, there are large amount of solid precipitated, was filtered, the filter cake washed once with 100ml methanol obtained 42.3g of yellow solid was purified by Technip Trinidad (I). Yield 93.6%.
ΧΗNMR (400MHz, dmso) δ11.94 (s, 1Η), 8.36 (s, 1H), 7.96 (dd, J = 11.9, 5.0Hz, 2H), 7.67 (d, J = 8.1Hz, 1H) , 7.16 (ddd, J = 26.9, 22.1, 7.0Hz, 5H), 6.85 (d, J = 8.6Hz, 2H), 6.61 (d, J = 8.7Hz, 2H), 3.90 (s, 3H), 2.99 ( s, 3H), 2.65 (s, 2H), 2.50-2.30 (m, 8H), 2.20 (s, 3H) MS:. m / z540 (m + 1) + square
PATENT
WO2016037514

(I) 2.30g, yield 85.3%. Melting point 241 ~ 243 ℃, Mass spectrum (the EI): m / Z 540 (the M + the H), 1 the H NMR (of DMSO D . 6 ): 2.27 (S, 3H), 2.43 (m, 8H), 2.78 (S, 2H) , 3.15 (s, 3H), 3.82 (s, 3H), 5.97 (d, J = 8.3Hz, 1H), 6.77 (d, J = 8.7Hz, 1H), 6.96 (d, J = 8.6Hz, 2H) , 7.32-7.62 (m, 8H), 8.15 (s, 1H), 12.15 (s, 1H).
CLIPS
http://pubs.rsc.org/en/content/articlelanding/2015/ay/c5ay01207d#!divAbstract
References
- Hilberg, F.; G. J. Roth, M. Krssak, S. Kautschitsch, W. Sommergruber, U. Tontsch-Grunt, P. Garin-Chesa, G. Bader, A. Zoephel, J. Quant, A. Heckel, W. J. Rettig (2008). “BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy”. Cancer Res 68 (12): 4774–82. doi:10.1158/0008-5472.CAN-07-6307. ISSN 1538-7445. PMID 18559524.
- Hilberg, F.; U. Tontsch-Grunt, F. Colbatzky, A. Heckel, R. Lotz, J.C.A. van Meel, G.J. Roth (2004). “BIBF1120 a novel, small molecule triple angiokinase inhibitor: profiling as a clinical candidate for cancer therapy”. European Journal of Cancer Supplements 2 (50).
- Reck, M.; R. Kaiser; C. Eschbach; M. Stefanic; J. Love; U. Gatzemeier; P. Stopfer; J. von Pawel (2011). “A phase II double-blind study to investigate efficacy and safety of two doses of the triple angiokinase inhibitor BIBF 1120 in patients with relapsed advanced non-small-cell lung cancer”. Ann Oncol. ISSN 1569-8041.
- Okamoto, I.; H. Kaneda, T. Satoh, W. Okamoto, M. Miyazaki, R. Morinaga, S. Ueda, M. Terashima, A. Tsuya, A. Sarashina, K. Konishi, T. Arao, K. Nishio, R. Kaiser, K. Nakagawa (2010). “Phase I safety, pharmacokinetic, and biomarker study of BIBF 1120, an oral triple tyrosine kinase inhibitor in patients with advanced solid tumors”. Mol Cancer Ther 9 (10): 2825–33. doi:10.1158/1535-7163.MCT-10-0379. ISSN 1538-8514. PMID 20688946.
- Mross, K.; M. Stefanic, D. Gmehling, A. Frost, F. Baas, C. Unger, R. Strecker, J. Henning, B. Gaschler-Markefski, P. Stopfer, L. de Rossi, R. Kaiser (2010). “Phase I study of the angiogenesis inhibitor BIBF 1120 in patients with advanced solid tumors”. Clin Cancer Res 16 (1): 311–9. doi:10.1158/1078-0432.CCR-09-0694. ISSN 1078-0432. PMID 20028771.
- Ledermann, J.A. (2009). “A randomised phase II placebo-controlled trial using maintenance therapy to evaluate the vascular targeting agent BIBF 1120 following treatment of relapsed ovarian cancer (OC)”. J Clin Oncol 27 (15s): (suppl; abstr 5501).
- Kropff, M.; J. Kienast; G. Bisping; W. E. Berdel; B. Gaschler-Markefski; P. Stopfer; M. Stefanic; G. Munzert (2009). “An open-label dose-escalation study of BIBF 1120 in patients with relapsed or refractory multiple myeloma”. Anticancer Res 29 (10): 4233–8. ISSN 1791-7530. PMID 19846979.
- Ellis, P. M.; R. Kaiser; Y. Zhao; P. Stopfer; S. Gyorffy; N. Hanna (2010). “Phase I open-label study of continuous treatment with BIBF 1120, a triple angiokinase inhibitor, and pemetrexed in pretreated non-small cell lung cancer patients”. Clin Cancer Res 16 (10): 2881–9. doi:10.1158/1078-0432.CCR-09-2944. ISSN 1078-0432. PMID 20460487.
- du Bois, A.; J. Huober; P. Stopfer; J. Pfisterer; P. Wimberger; S. Loibl; V. L. Reichardt; P. Harter (2010). “A phase I open-label dose-escalation study of oral BIBF 1120 combined with standard paclitaxel and carboplatin in patients with advanced gynecological malignancies”. Ann Oncol 21 (2): 370–5. doi:10.1093/annonc/mdp506. ISSN 1569-8041. PMID 19889612.
- Xiang, Q. F.; F. Wang; X. D. Su; Y. J. Liang; L. S. Zheng; Y. J. Mi; W. Q. Chen; L. W. Fu (2011). “Effect of BIBF 1120 on reversal of ABCB1-mediated multidrug resistance”. Cell Oncol (Dordr) 34 (1): 33–44. doi:10.1007/s13402-010-0003-7. ISSN 2211-3436.
- “Boehringer Ingelheim – AGO-OVAR 12 / LUME-Ovar 1 Trial Information”. 2011.
- “Boehringer Ingelheim – LUME-Lung 2 Trial Information”. 2011.
- “Boehringer Ingelheim – LUME-Lung 1 Trial Information”. 2011.
- http://clinicaltrials.gov/ct2/results?term=++%09+BIBF+1120&phase=1
- http://clinicaltrials.gov/ct2/show/NCT00805194 Phase III LUME-Lung 1: BIBF 1120 Plus Docetaxel as Compared to Placebo Plus Docetaxel in 2nd Line Non Small Cell Lung Cancer
- http://clinicaltrials.gov/ct2/show/NCT01015118 Phase III BIBF 1120 or Placebo in Combination With Paclitaxel and Carboplatin in First Line Treatment of Ovarian Cancer
- http://clinicaltrials.gov/ct2/show/NCT01335477 Safety and Efficacy of BIBF 1120 at High Dose in Idiopathic Pulmonary Fibrosis Patients II
- “FDA approves Ofev to treat idiopathic pulmonary fibrosis”. 2014.
-
F. Hilberg et al. Cancer Res. 2008, 68, 4774
2. M. Reck et al. Ann. Oncol. 2011, 22, 1374
3. M. Reck et al. J. Clin. Oncol. 2013 (suppl.), Abst LBA8011
4. N. H. Hanna et al. J. Clin. Oncol. 2013, 2013 (suppl.), Abst 8034
5. J.A. Ledermann et al. J. Clin Oncol. 2011, 29, 3798
6. Glioblastoma: A. Muhac et al. J. Neurooncol. 2013, 111, 205
7. O. Bouche et al. Anticancer Res. 2011, 31, 2271
8. T. Eisen et al. J. Clin. Oncol. 2013 (suppl.), Abst. 4506
MORE…………….
Reference:
[6]. Japan PMDA.
[7]. Drug@FDA, NDA205832 Pharmacology Review(s).
[8]. Med. Chem. 2015, 58, 1053-1063.
[9]. Drug@EMA, EMEA/H/C/002569 Vargatef: EPAR-Assessment Report.
[10]. Drug Des. Devel. Ther. 2015, 9, 6407-6419.
[11]. Cancer Res. 2008, 68, 4774-4782.
[12]. J. Med. Chem. 2009, 52, 4466-4480.
[13]. J. Pharmacol. Exp. Ther. 2014, 349, 209-220.
[14]. Clin. Cancer. Res. 2015, 21, 4856-4867.
Merten, J.; et. al. Process for the manufacture of an indolinone derivative. US20110201812A1
2. Roth, G. J.; et. al. 3-z-[1-(4-(n-((4-methyl-piperazin-1-yl)-methylcarbonyl)-n-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone-monoethanesulphonate and the use thereof as a pharmaceutical composition. WO2004013099A1
3. Roth, G. J.; et. al. Design, Synthesis, and Evaluation of Indolinones as Triple Angiokinase Inhibitors and the Discovery of a Highly Specific 6-Methoxycarbonyl-Substituted Indolinone (BIBF 1120). J Med Chem, 2009, 52(14), 4466-4480.
-

ニンテダニブエタンスルホン酸塩
Nintedanib Ethanesulfonate
C31H33N5O4.C2H6O3S : 649.76
[656247-18-6] |
| US7119093 * |
Jul 21, 2003 |
Oct 10, 2006 |
Boehringer Ingelheim Pharma Gmbh & Co. Kg |
3-Z-[1-(4-(N-((4-Methyl-piperazin-1-yl)-methylcarbonyl)-N-methyl-amino)-anilino)-1-phenyl-methylene]-6-methoxycarbonyl-2-indolinone-monoethanesulphonate and the use thereof as a pharmaceutical composition |
///////////////












.jpg)


 Highlights
Highlights






















.jpg)
































 COMPD1
COMPD1