











 .
.
TRICYCLOHEXADECAHEXAENE DERIVATIVES FOR USE IN THE TREATMENT OF HEPATITIS C VIRUS
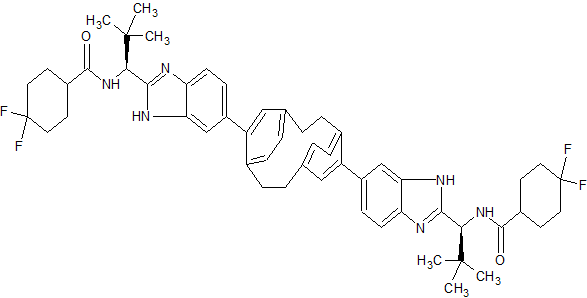
WO2015026454, COMBINATIONS COMPRISING TRICYCLOHEXADECAHEXAENE DERIVATIVES FOR USE IN THE TREATMENT OF HEPATITIS C VIRUS
BRISTOL-MYERS SQUIBB COMPANY [US/US]; Route 206 and Province Line Road Princeton, New Jersey 08543 (US)
PATENT WO2015026454 [LINK]
WANG, Alan Xiangdong; (US).
LOPEZ, Omar D.; (US).
TU, Yong; (US).
BELEMA, Makonen; (US)
Example B-l
Example B-l Step a

To a solution of 4-bromobenzene-l,2-diamine (2.5 g, 13.37 mmol) in DCM (30 mL) was added (S)-2-((tert-butoxycarbonyl)amino)-3,3-dimethylbutanoic acid (3.09 g, 13.37 mmol), DIPEA (2.334 mL, 13.37 mmol) and HATU (5.08 g, 13.37 mmol). The reaction mixture was stirred at room temperature for 18 h. The reaction mixture was diluted with water and extracted with DCM. The organic phase was washed with brine, dried over Na2S04, filtered and concentrated. The crude material was purified by ISCO using 40 g Redisep silica column, CHCl3/MeOH as eluant to obtain (S)-tert-butyl ( 1 -((2-amino-4-bromophenyl)amino)-3 ,3 -dimethyl- 1 -oxobutan-2-yl) carbamate (1.82 g) as yellow solid. LC (Condition 1): Rt = 2.13 min. LC/MS: Anal. Calcd. for [M+H20]+ Ci7H27BrN204 : 402.12; found 402.2. 1H NMR (DMSO-d6, δ = 2.50 ppm, 400 MHz): δ 9.35 – 9.21 (m, 1 H), 7.07 (d, J= 8.5 Hz, 1 H), 6.91 (d, J= 2.0 Hz, 1 H), 6.80 – 6.60 (m, 1 H), 5.25 – 5.01 (m, 2 H), 4.07 – 3.89 (m, 1 H), 1.52 – 1.34 (m, 9 H), 1.02 – 0.86 (m, 9 H).
Example B-l, Step b

Acetic acid (15 mL) was added to (S)-tert-butyl (l-((2-amino-4-bromo phenyl)amino)-3,3-dimethyl-l-oxobutan-2-yl)carbamate (1.8 g, 4.50 mmol) and the reaction mixture was heated to 65 °C for overnight. The volatile component was removed in vacuo, and the residue was co-evaporated with dry CH2C12 (2 x 15 mL). The organic phase was washed with saturated NaHC03 solution, brine, dried over Na2S04 and concentrated to obtain (S)-tert-butyl (l-(6-bromo-lH-benzo[d] imidazol-2-yl)-2,2-dimethyl propyl)carbamate (1.68 g) as yellow solid. LC (Condition 1): Rt = 2.19 min. LC/MS: Anal. Calcd. for [M+H]+ Ci7H25BrN302 : 381.11; found 382.2. 1H NMR (DMSO-dg, δ = 2.50 ppm, 300 MHz): δ 12.46 – 12.27 (m, 1 H), 7.82 – 7.65 (m, 1 H), 7.59 – 7.41 (m, 1 H), 7.29 (dt, J= 1.9, 8.5 Hz, 1 H), 7.12 – 6.90 (m, 1 H), 4.64 (d, J= 9.8 Hz, 1 H), 1.44 – 1.27 (m, 9 H), 0.88 (br. s., 9 H).
-1 Step c

To a solution of (S)-tert-butyl (l-(6-bromo-lH-benzo[d]imidazol-2-yl)-2,2-dimethyl propyl) carbamate (1.57 g, 4.11 mmol) in dioxane (25 mL) was added bis (pinacolato)diboron (1.564 g, 6.16 mmol) and potassium acetate (1.209 g, 12.32 mmol). The reaction mixture was purged with argon for 10 min then PdCl2(dppf) (0.150 g, 0.205 mmol) was added to the above reaction mixture and again purged with argon for 5 min. The reaction mixture was heated to 90 °C for overnight. The reaction mixture was diluted with water (15 ml) and extracted with EtOAc (2 x 25 ml). The combined organic phase was washed with brine, dried over Na2S04 and concentrated in vacuo. The crude material was purified by ISCO using 40 g Redisep column, hexane/ethyl acetate as eluant to afford (S)-tert-butyl (2,2-dimethyl-l-(6-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 H-benzo[d]imidazol-2-yl)propyl) carbamate (1.35 g) as yellow solid. LC (Condition 1): Rt = 2.21 min. LC/MS: Anal. Calcd. for [M+H]+ C23H37BN304 : 430.29; found 430.4. 1H NMR (CD3OD, δ = 3.34 ppm, 400 MHz): δ 7.98 (s, 1 H), 7.65 (dd, J= 1.0, 8.5 Hz, 1 H), 7.53(d, J= 8.5 Hz, 1 H), 4.73 (br. s., 1 H), 1.37 (s, 12 H), 1.24 (m, 9 H), 1.01 (s, 9 H).
-1 Step d

To a solution of (S)-tert-butyl (2,2-dimethyl-l-(6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-benzo[d]imidazol-2-yl)propyl)carbamate (1.114 g, 2.59 mmol) and 4,16-dibromo[2,2]paracyclophane (0.38g, 1.038 mmol) in dioxane (10 mL) was added Cs2C03 (0.845 g, 2.59 mmol) in water (2 mL) and degassed for 10 min.
PdCl2(dppf) (0.038 g, 0.052 mmol) was added to the above reaction mixture and again degassed for 5 min. The reaction mixture was heated to 90 °C for 12 h. Then the reaction mixture was filtered to get Example B-1 Step d which was taken for next step without further purification. LC (Condition 1): Rt = 2.54 min. LC/MS: Anal. Calcd. for [M+H]+ ^0Η63Ν6Ο4 : 811.49; found 811.6. 1H NMR (DMSO-d6, δ = 2.50 ppm, 300 MHz): δ 12.36 (br. s., 2 H), 7.85 – 7.52 (m, 4 H), 7.32 (d, J= 7.9 Hz, 2 H), 7.05 (br. s., 2 H), 6.89 – 6.67 (m, 4 H), 6.54 (br. s., 2 H), 4.72 (d, J= 8.7 Hz, 2 H), 3.57 – 3.44 (m, 2 H), 3.07 (br. s., 2 H), 2.83 (br. s., 2 H), 2.65 (br. s., 2 H), 1.36 (s, 18 H), 1.08 – 0.91 (m, 18 H).
-1 Step e

HC1 in dioxane (4 mL, 24.00 mmol) was added to Example B-1 Step d (0.1 g,
0.102 mmol), and the reaction mixture was allowed to stir at RT for 2 h. Completion of the reaction was monitored by LCMS. The volatile component was removed in vacuo and the residue was washed with diethyl ether and dried to afford Example B-1 Step e (0.07 g) as yellow solid. LC (Condition 1): R, = 2.54 min. LC/MS: Anal.
Calcd. for [M+H]+ C40H47N6 : 611.39; found 611.4. 1H NMR (CD3OD, δ = 3.34 ppm, 400 MHz): δ 7.90 (d, J= 13.1 Hz, 2 H), 7.83 (d, J= 8.5 Hz, 2 H), 7.61 (d, J= 8.5 Hz, 2 H), 6.84 (d, J= 6.5 Hz, 2 H), 6.78 (s, 2 H), 6.70 – 6.65 (m, 2 H), 4.54 (d, J= 1.0 Hz, 2 H), 3.54 – 3.46 (m, 2 H), 3.18 – 3.10 (m, 2 H), 2.98 – 2.86 (m, 2 H), 2.71 (br. s., 2 H), 1.25 – 1.22 (m, 18 H).
To a solution of Example B-1 Step e (0.04 g, 0.053 mmol) in DMF (5 mL) was added 4,4-difluorocyclohexanecarboxylic acid (0.017 g, 0.106 mmol), DIPEA (0.055 mL, 0.317 mmol) and HATU (0.030 g, 0.079 mmol). After being stirred for 2 h at room temperature, the volatile component was removed in vacuo and the residue was dissolved in DCM (10 mL), washed with saturated solution of NH4C1, 10% NaHC03 solution, brine, dried over Na2S04 and concentrated in vacuo. The crude was purified by reverse phase HPLC purification to give Example B-1 as a white solid. LC (Condition 1): R, = 2.37 min. LC/MS: Anal. Calcd. for [M+H]+
C54H63F4N602: 903.49; found 903.4. 1H NMR (DMSO-d6, δ = 2.50 ppm, 400 MHz): δ 12.53 – 12.32 (m, 2 H), 8.41 – 8.21 (m, 2 H), 7.84 – 7.50 (m, 4 H), 7.43 – 7.24 (m, 2 H), 6.90 – 6.67 (m, 4 H), 6.60 – 6.44 (m, 2 H), 5.14 – 4.97 (m, 2 H), 3.44 (br. s., 2 H), 3.08 (br. s., 2 H), 2.93 – 2.77 (m, 2 H), 2.73 – 2.56 (m, 4 H), 2.20 – 1.98 (m, 3 H), 1.96 – 1.49 (m, 13 H), 1.02 (s, 18 H).
Starting materials can be obtained from commercial sources or prepared by well-established literature methods known to those of ordinary skill in the art. Acid precursors for the final step can be prepared according to the methods described in U.S. Patent Application Serial No. 13/933495, filed July 2, 2013.
LC/MS Condition 1
Column = Ascentis Express C18, 2.1 X 50 mm, 2.7 um
Solvent A = CH3CN (2%) + 10 mM NH4COOH in H20 (98%)
Solvent B = CH3CN (98%) + 10 mM NH4COOH in H20 (2%)
Start %B = 0; Final %B = 100
Gradient time = 1.4 min; Stop time = 4 min
Stop time = 4 min
Flow Rate = 1 mL/min; Wavelength = 220 nm
LC/MS Condition 2
Column = Waters BEH CI 8, 2.0 x 50 mm, 1.7 μιη
Slovent A = ACN (5%) + H20 (95%) containing 10 mM NH4OAc
Solvent B = ACN (95%) + H20 (5%) containing 10 mM NH4OAc
Start %B = 0; Final %B = 100
Gradient time = 3 min
Flow Rate = 1 mL/min
Wavelength = 220 nm
Temperature = 50 °C
LC/MS Condition 3
Column: Waters Phenomenex CI 8, 2.0 x 30 mm, 3 μιη particle
Mobile Phase A: 10% MeOH:90% Water :0.1%TFA
Mobile Phase B: 90% MeOH: 10% Water :0.1%TFA
Gradient: 0%B, 0-100% B over 3 minutes, then a 1 -minute hold at 100% B Flow: 0.8mL/min
Detection: 220 nm
Temperature: 40 °C
LC/MS Condition 4
Column: Waters BEH CI 8, 2.0 x 50 mm, 1.7 μιη particle
Mobile Phase A: 5:95 acetonitrile: water with 10 mM ammonium acetate Mobile Phase B: 95:5 acetonitrile: water with 10 mM ammonium acetate Gradient: 0%B, 0-100% B over 3 minutes, then a 0.5-minute hold at 100% B Flow: 1 mL/min
Detection: UV at 220 nm
Temperature: 50 °C
/////////
FC1(F)CCC(CC1)C(=O)N[C@H](c2nc3ccc(cc3n2)c9cc4ccc9CCc5ccc(CC4)c(c5)c6ccc7nc(nc7c6)[C@@H](NC(=O)C8CCC(F)(F)CC8)C(C)(C)C)C(C)(C)C

Sorry, the comment form is closed at this time.