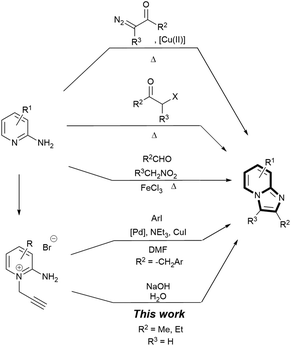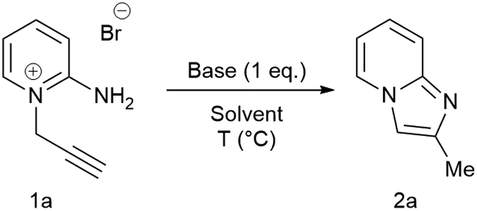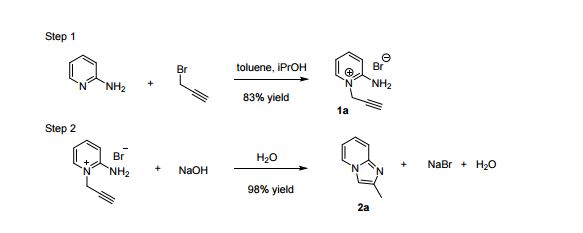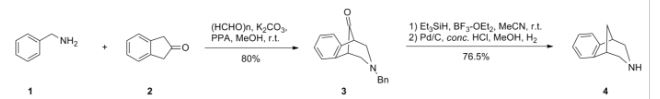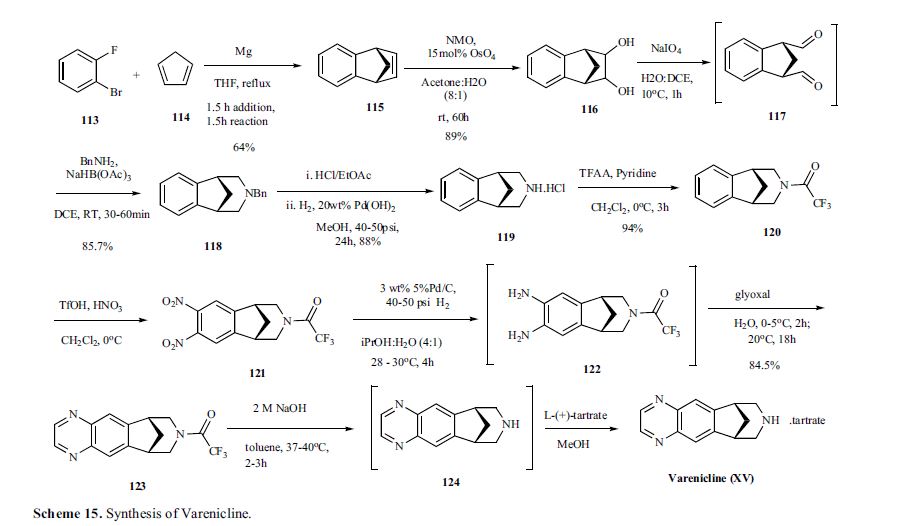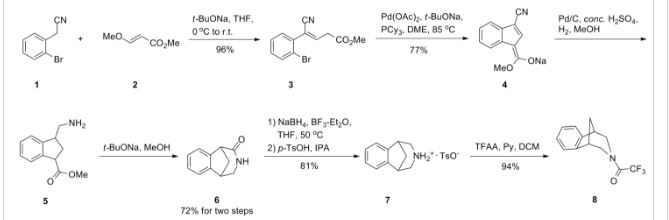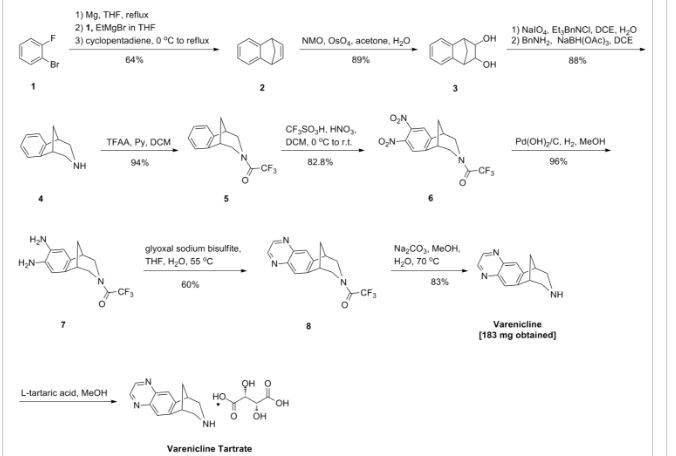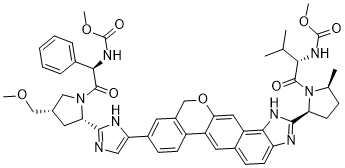
VELPATASVIR (GS-5816), GILEAD SCIENCES
CAS 1377049-84-7
| Molecular Formula: |
C49H54N8O8 |
| Molecular Weight: |
883.00186 g/mol |
Hepatitis C virus NS 5 protein inhibitors
KEEP WATCHING AS I ADD MORE DATA, SYNTHESIS……………
Gilead Sciences, Inc. INNOVATOR
Elizabeth M. Bacon, Jeromy J. Cottell, Ashley Anne Katana, Darryl Kato, Evan S. Krygowski, John O. Link, James Taylor, Chinh Viet Tran, Martin Teresa Alejandra Trejo, Zheng-Yu Yang, Sheila Zipfel,

Senior Research Associate II at Gilead Sciences

Methyl {(2S)-1-[(2S,5S)-2-(5-{2-[(2S,4S)-1-{(2R)-2- [(methoxycarbonyl)amino]-2-phenylacetyl}-4- (methoxymethyl)pyrrolidin-2-yl]-1 ,1 1 dihydroisochromeno[4′,3′:6,7]naphtho[1 ,2-d]imidazol-9-yl}-1 H-imidazol-2-yl)-5- methylpyrrolidin-1 -yl]-3-methyl-1 -oxobutan-2-yl}carbamate
methyl {(2S)-1-[(2S,5S)-2-(9-{2-[(2S,4S)-1-{(2R)-2-[(methoxycarbonyl)amino]-2-phenylacetyl}-4-(methoxymethyl)pyrrolidin-2-yl]-1H-imidazol-5-yl}-1,11-dihydroisochromeno[4′,3′:6,7]naphtho[1,2-d]imidazol-2-yl)-5-methylpyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl}carbamate
methyl {(2S)-1 – [(2S,5S)-2-(5-{2-[(2S,4S)-l- {(2R)-2-[(methoxycarbonyl)amino]-2-phenylacetyl} -4-(methoxymethyl) pyrrolidin-2-yl]-l,l 1 dihydroisochromeno [4′,3′:6,7]naphtho[l,2-d]imidazol-9-yl}-lH-imidazol-2-yl)- 5-methylpyrrolidin-l-yl]-3-methyl-l -oxobutan-2-yl}carbamate

Research Scientist I at Gilead Sciences
{(2S)-1-[(2S,5S)-2-(9-{2-[(2S,4S)-1-{(2R)-2-[(Méthoxycarbonyl)amino]-2-phénylacétyl}-4-(méthoxyméthyl)-2-pyrrolidinyl]-1H-imidazol-4-yl}-1,11-dihydroisochroméno[4′,3′:6,7]naphto[1,2-d]imidazol-2-yl)-5 -méthyl-1-pyrrolidinyl]-3-méthyl-1-oxo-2-butanyl}carbamate de méthyle
Carbamic acid, N-[(1R)-2-[(2S,4S)-2-[4-[1,11-dihydro-2-[(2S,5S)-1-[(2S)-2-[(methoxycarbonyl)amino]-3-methyl-1-oxobutyl]-5-methyl-2-pyrrolidinyl][2]benzopyrano[4′,3′:6,7]naphth[1,2-d]imidazol-9-yl]-1H- imidazol-2-yl]-4-(methoxymethyl)-1-pyrrolidinyl]-2-oxo-1-phenylethyl]-, methyl ester
Methyl {(2S)-1-[(2S,5S)-2-(9-{2-[(2S,4S)-1-{(2R)-2-[(methoxycarbonyl)amino]-2-phenylacetyl}-4-(methoxymethyl)pyrrolidin-2-yl]-1H-imidazol-4-yl}-1,11-dihydro[2]benzopyrano[4′,3′:6,7]naphtho[1,2-d]imidazol-2-yl)-5-methylpyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl}carbamate




 .
.

| Description |
Pan-genotypic HCV NS5A inhibitor |
| Molecular Target |
HCV NS5A protein |
| Mechanism of Action |
HCV non-structural protein 5A inhibitor |
| Therapeutic Modality |
Small molecule |
| Latest Stage of Development |
Phase II |
| Standard Indication |
Hepatitis C virus (HCV) |
| Indication Details |
Treat HCV genotype 1 infection; Treat HCV infection |
- Gilead Sciences
- Class Antivirals; Carbamates; Chromans; Imidazoles; Naphthols; Phenylacetates; Phosphoric acid esters; Pyrimidine nucleotides; Pyrrolidines; Small molecules
- Mechanism of Action Hepatitis C virus NS 5 protein inhibitors
Most Recent Events
- 14 Jul 2016 Registered for Hepatitis C in Canada (PO)
- 08 Jul 2016 Registered for Hepatitis C in Liechtenstein, Iceland, Norway, European Union (PO)
- 30 Jun 2016 Gilead Sciences plans a phase III trial for Hepatitis C (Combination therapy, Treatment-experienced) in Japan (PO (NCT02822794)

Darryl Kato works on a hepatitis treatment at Gilead Sciences Inc.’s lab
Velpatasvir, also known as GS-5816, is a potent and selective Hepatitis C virus NS5A inhibitor. GS-5816 has demonstrated pan-genotypic activity and a high barrier to resistance in HCV replicon assays. GS-5816 demonstrated pangenotypic antiviral activity in patients with genotype 1-4 HCV infection. It will be further evaluated in combination with other pangenotypic direct-acting antivirals to achieve the goal of developing a well-tolerated, highly effective treatment for all HCV genotypes.
|
WO 2013/075029. Compound I has the formula:
|

methyl {(2S)-1-[(2S,5S)-2-(9-{2-[(2S,4S)-1-{(2R)-2-[(methoxycarbonyl)amino]-2-phenylacetyl}-4-(methoxymethyl)pyrrolidin-2-yl]-1H-imidazol-5-yl}-1,11-dihydroisochromeno[4′,3′:6,7]naphtho[1,2-d]imidazol-2-yl)-5-methylpyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl}carbamate
PAPER
Patent Highlights: Recently Approved HCV NS5a Drugs
Cidara Therapeutics, 6310 Nancy Ridge Dr., Suite 101, San Diego, California 92121, United States
Org. Process Res. Dev., Article ASAP
Abstract
Five inhibitors of the NS5a enzyme have been approved as part of oral regimens for the treatment of hepatitis C virus, including daclatasvir (Bristol-Myers Squibb), ledipasvir (Gilead Sciences), ombitasvir (AbbVie), elbasvir (Merck), and velpatasvir (Gilead Sciences). This article reviews worldwide patents and patent applications that have been published on synthetic routes and final forms for these five drugs.
PATENT
https://google.com/patents/WO2013075029A1?cl=en
Example NP
Methyl {(2S)-1-[(2S,5S)-2-(5-{2-[(2S,4S)-1-{(2R)-2- [(methoxycarbonyl)amino]-2-phenylacetyl}-4- (methoxymethyl)pyrrolidin-2-yl]-1 ,1 1 dihydroisochromeno[4′,3′:6,7]naphtho[1 ,2-d]imidazol-9-yl}-1 H-imidazol-2-yl)-5- methylpyrrolidin-1 -yl]-3-methyl-1 -oxobutan-2-yl}carbamate
Methyl {(2S)-l-[(2S,5S)-2-(5-{2-[(2S,4S)-l-{(2R)-2-[(methoxycarbonyl)amino]-2-phenylacetyl}-4- (methoxymethyl)pyrrolidin-2-yl]-l,ll dihydroisochromeno [4′,3′:6,7]naphtho[l,2-d]imidazol-9- yl}-lH-imidazol-2-yl)-5-methylpyrrolidin-l-yl]-3-methyl-l-oxobutan-2-yl}carbamate
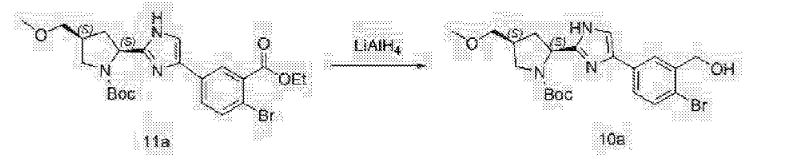
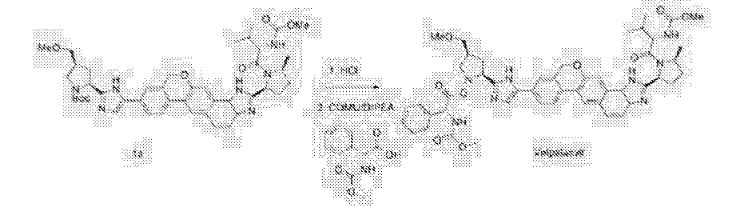
The synthesis of this compound was prepared according to the procedure of example LR-1 with the following modification. During the Suzuki coupling, (2S)-l-[(2S,5S)-2-(5-iodo-lH-imidazol- 2-yl)-5-methylpyrrolidin-l-yl]-2-[(l-meth^ was used in lieu of
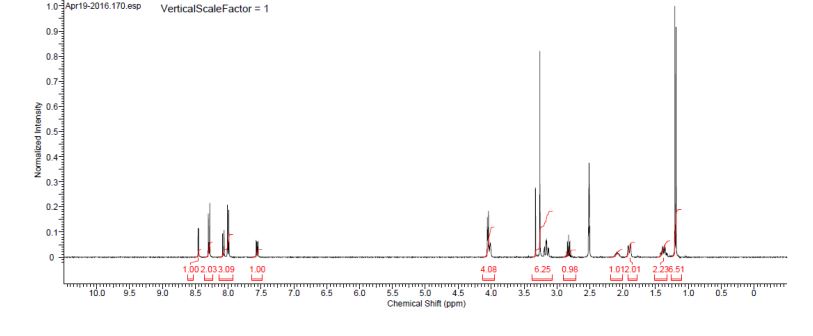
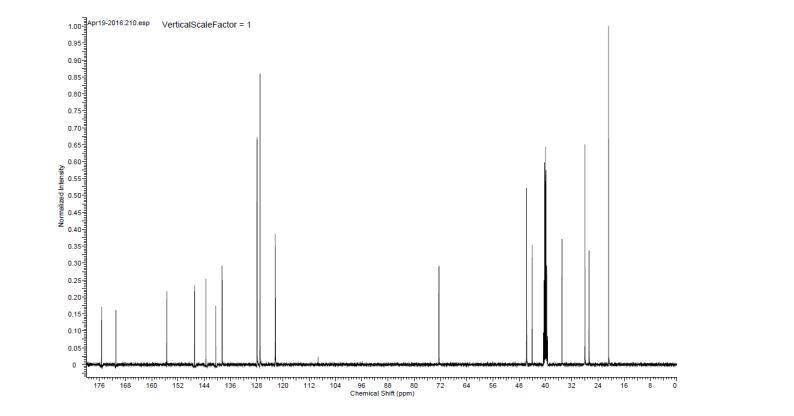
(2S)-l -[(2S)-2-(5-bromo-lH-imidazol-2-yl)pyrrolidin-l-yl]-2-[(l-methoxyethenyl)amino]-3- methylbutan-l-one. The crade material was purified by preparative HPLC to provide methyl {(2S)-1 – [(2S,5S)-2-(5-{2-[(2S,4S)-l- {(2R)-2-[(methoxycarbonyl)amino]-2-phenylacetyl} -4-(methoxymethyl) pyrrolidin-2-yl]-l,l 1 dihydroisochromeno [4′,3′:6,7]naphtho[l,2-d]imidazol-9-yl}-lH-imidazol-2-yl)- 5-methylpyrrolidin-l-yl]-3-methyl-l -oxobutan-2-yl}carbamate as a white solid (17 mg, 0.019 mmol, 17%). lU NMR (400 MHz, cd3od) δ 8.63 (s, 1H), 8.19 (d, 1H), 8.04 (m, 1H), 7.87 (m, 2H), 7.66 (m, 2H), 7.52 – 7.39 (m, 6H), 5.50 (m, 2H), 5.32 (s, 2H), 5.16 (m, 1H), 4.12 (m, 1H), 3.80 (m, 4H), 3.66 (s, 6H), 3.43 (m, 4H), 3.23 (s, 3H), 2.72-1.99 (m, 9H), 1.56 (d, 3H), 1.29 (m, 1H), 0.99 (d, 3H), 0.88 (d, 3H).
PATENT
US 20150361073 A1
Scheme 1
Compound (J)
 Compound (I) H CO- Com pound (G)
Compound (I) H CO- Com pound (G)


st alkylation: Conversion of Compound (I-a) to Compound (G-a)

Compound (I-a) (45 g, 1.0 equiv.), Compound (J-a) (26.7g, 1.03 equiv.) and potassium carbonate (20.7g, 1.5 equiv.) in dichloromethane (450 mL) were stirred at about 20 °C for approximately 3-4 hours. After the completion of the reaction, water (450 mL) was charged into the reactor and the mixture was stirred. Layers were separated, and the aqueous layer was extracted with dichloromethane (200 mL). The combined organic layers were washed with 2 wt% NaH2PO4/10wt% NaCl solution (450 mL). The organic layer was then concentrated and the solvent was swapped from dichloromethane into tetrahydrofuran. A purified sample of Compound (G-a) has the following spectrum: ¾ NMR (400 MHz,
CDC13) δ 7.90-7.94 (m, 1H), 7.81-7.85 (m, 1H), 7.72 (s, 1H), 7.69 (s, 1H), 7.66 (s, 1H), 5.19-5.56 (2dd, 2H), 5.17 (s, 2H), 4.73 (t, 1H), 4.39-4.48 (m, 1H), 3.70-3.77 (m, 1H), 3.37-3.45 (m, 2H), 3.33-3.35 (d, 3H), 3.28-3.32 (m, 1H), 3.20-3.25 (dd, 1H), 2.92-2.96 (dt, 1H), 2.44-2.59 (m, 4H), 1.97-2.09 (m, 1H), 1.44 (d, 9H).
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, alternative starting material may be Compound (I) where X may be -CI, -Br, -OTs, -OS02Ph, -OS02Me, -OS02CF3, -OS02R, , and -OP(0)(OR)2 and Y may be -CI, -Br, -OTs, -OS02Ph, -OS02Me, -OS02CF3, -OS02R, and -OP(0)(OR)2. R may be alkyl, haloalkyl, or an optionally substituted aryl.
Various bases may also be employed, such as phosphate salts (including but not limited to KH2P04, K3P04, Na2HP04, and Na3P04) and carbonate salts (including but not limited to Na2C03,Cs2C03, and NaHC03). Where the starting material is Compound (J), KHC03 or preformed potassium, sodium, and cesium salts of Compound (J) may also be used.
Alternative solvents can include 2-methyltetrahydrofuran, tetrahydrofuran, isopropyl acetate, ethyl acetate, tert-butyl methyl ether, cyclopentyl methyl ether, dimethylformamide, acetone, MEK, and MIBK.
The reaction temperature may range from about 10 °C to about 60 °C.
” alkylation: Conversion of Compound (G-a) to Compound (B-a):

A solution of Compound (G-a) (prepared as described earlier starting from 45 g of Compound (I-a)) was mixed with Compound (H) (42.9g, 1.5 equiv.), and cesium carbonate (26. lg, 0.8 equiv.). The reaction mixture was stirred at about 40-45 °C until reaction was complete and then cooled to about 20 °C. Water (450 mL) and ethyl acetate (225 mL) were added and the mixture was agitated. Layers were separated, and the aqueous layer was extracted with ethyl acetate (150 mL). Combined organic phase was concentrated and solvent was swapped to toluene. A purified sample of Compound (B-a) has the following spectrum: ¾ NMR (400 MHz, CDC13) 57.90-7.93 (m, 1H), 7.81-7.83 (m, 1H), 7.73 (s, 1H), 7.63-7.64 (d, 1H), 7.59-7.60 (d, 1H), 5.52-5.63 (m, 1H), 5.30-5.43 (q, 1H), 5.13-5.23 (s+m, 3H), 4.56-4.64 (m, 2H), 4.39-4.48 (m, 1H), 4.20-4.27 (m, 1H), 3.62-3.79 (m, 2H), 3.66 (s, 2H), 3.36-3.45 (m, 2H), 3.34-3.35 (d, 3H), 3.07-3.25 (m, 3H), 2.59-2.37 (m, 5H), 1.97-2.16 (m, 3H), 1.60 (s, 3H), 1.38-1.45 (m, 12H), 0.91-1.03 (m, 6H).
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, alternative starting material may include Compound (G) where Y may be -CI, -Br, -OTs, -OS02Ph, -OS02Me, -OS02CF3, -OS02R, , or -OP(0)(OR)2. where R is alkyl, aryl, or substituted aryl. In some embodiments, the substituted aryl may be an aryl having one or more substituents, such as alkyl, alkoxy, hydroxyl, nitro, halogen, and others as discussed above.
Various bases may be employed. Non-limiting examples can include phosphate salts (including but not limited to KH2P04, K3P04, Na2HP04, and Na3P04) and carbonate salts (including but not limited to K2C03 or Na2C03). If Compound (H) is used as the starting material, Li2C03 or preformed potassium, sodium, and cesium salt of Compound (H) may be employed.
Alternative solvents may include 2-methyltetrahydrofuran, dichloromethane, toluene, mixtures of THF/Toluene, isopropyl acetate, ethyl acetate, l-methyl-2-pyrrolidinone, Ν,Ν-dimethylacetamide, acetone, MEK,and MIBK. An alternative additive may be
potassium iodide, and the reaction temperature may range from about 40 °C to about 60 °C or about 40 °C to about 50 °C.

A toluene solution of Compound (B-a) (604 g solution from 45 g of Compound (I-a)) was charged to a reaction vessel containing ammonium acetate (185.2 g) and isopropanol (91.0 g). The contents of the reactor were agitated at about 90 °C until the reaction was complete (about 16 to 24 hours). The reaction mixture was cooled to about 45 °C, and then allowed to settle for layer separation. Water (226 g) was added to the organic phase, and the resulting mixture was separated at about 30 °C. Methanol (274 g), Celite (26.9 g) and an aqueous solution of sodium hydroxide (67.5 g, 50%) and sodium chloride (54.0 g) in water (608 g) were added to the organic phase, and the resulting mixture was agitated for a minimum of 30 minutes. The mixture was then filtered through Celite and rinsed forward with a mixture of toluene (250 g) and isopropanol (1 1 g). The biphasic filtrate was separated and water (223 g) was added to the organic phase, and the resulting mixture was agitated at about 30 °C for at least 15 minutes. The mixture was filtered through Celite and rinsed forward with toluene (91 g). The organic layer was concentrated by vacuum distillation to 355 g and was added over 30 minutes to another reactor containing w-heptane (578 g). The resulting slurry is filtered, with the wetcake was washed with w-heptane (450 mL) and dried in a vacuum oven to afford Compound (C-a). A purified sample of Compound (C-a) has the following spectrum: *H NMR (400 MHz, CDC13) δ 12.27-11.60 (m, 1 H), 1 1.18-10.69 (m, 1 H), 7.83 – 7.44 (m, 4 H), 7.36 (d, J = 7.9 Hz, 1 H), 7.28 – 7.05 (m, 1 H), 5.65 – 5.25 (m, 1H), 5.25 – 4.83 (m, 4 H), 4.34 – 4.03 (m, 2 H), 3.93 – 3.63 (m, 4 H), 3.52 (s, 1 H), 3.35 (d, J = 2.4 Hz, 4 H), 3.19 – 2.94 (m, 4 H), 2.88 (dd, J = 12.0, 7.9 Hz, 3 H), 2.66 – 1.85 (m, 5 H), 1.79 (s, 5 H), 1.37 – 1.12 (m, 6H), 1.04-0.98 (m, 6 H), 0.82 (t, J = 7.7 Hz, 2 H).
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, alternative reagents, in lieu of ammonium acetate, can include hexamethyldisilazane, ammonia, ammonium formate, ammonium propionate, ammonium hexanoate, and ammonium octanoate. Various solvents, such as toluene, xylene, an alcohol
(including but not limited to isopropanol, 1-propanol, 1-butanol, 2-butanol, 2-methoxyethanol, and glycols, such as ethylene glycol and propylene glycol) may be employed. Alternative catalyst/additives may include magnesium stearate, acetic acid, propionic acid, and acetic anhydride. The reaction temperature may range from about 60 °C to about 110 °C or about 85 °C to about 95 °C.
D

Preparation of Compound (D-a) using DDQ as oxidant:
A solution of Compound (C-a) (255.84 g) in 2-methyltetrahydrofuran (1535 mL) was cooled to about 0 °C and acetic acid (0.92 mL) was added. To this mixture was added a solution of DDQ (76.98 g) in 2-methyltetrahydrofuran (385 mL) over about 30 minutes. Upon reaction completion, a 10 wt% aqueous potassium hydroxide solution (1275 mL) was added over about 30 minutes and the mixture was warmed to about 20 °C. Celite (101.5 g) was added and the slurry was filtered through Celite (50.0 g) and the filter cake was rinsed with 2-methyltetrahydrofuran (765 mL). The phases of the filtrate were separated. The organic phase was washed successively aqueous potassium hydroxide solution (1020 mL, 10 wt%), aqueous sodium bisulfite solution (1020 mL, 10 wt%), aqueous sodium bicarbonate solution (1020 mL, 5 wt%) and aqueous sodium chloride solution (1020 mL, 5 wt%). The organic phase was then concentrated to a volume of about 650 mL. Cyclopentyl methyl ether (1530 mL) was added and the resulting solution was concentrated to a volume of about 710 mL. The temperature was adjusted to about 40 °C and Compound (D-a) seed (1.0 g) was added. The mixture was agitated until a slurry forms, then methyl tert-butyl ether (2300 mL) was added over about 3 hours. The slurry was cooled to about 20 °C over about 2 hours and filtered. The filter cake was rinsed with methyl tert-butyl ether (1275 mL) and dried in a vacuum oven at about 40 °C to provide Compound (D-a). A purified sample of Compound (D-a) has the following spectrum: ¾ NMR (400 MHz, CDC13) δ 13.05-10.50 (comp m, 2H), 8.65-6.95 (comp m, 8H), 5.50-5.35 (m, 2H), 5.25^1.60 (comp m, 3H), 4.35-4.20 (m, 1H), 4.00-3.65 (comp m, 4H), 3.60-3.45 (m, 1H), 3.45-3.25 (comp m, 4H), 3.25-3.00 (comp m, 2H), 2.95-1.65 (comp m, 6H), 1.47 (br s, 9H), 1.40-1.25 (comp m, 2H), 1.20-0.70 (comp m, 9H).
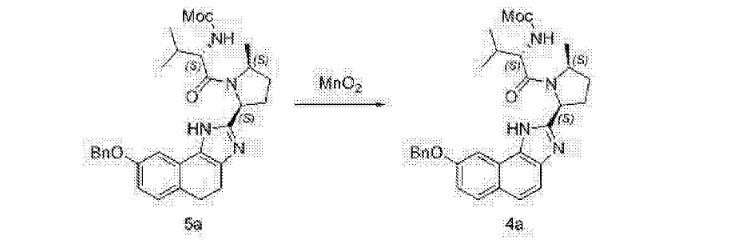
Alternative Preparation of Compound (D-a) using Mn02 as oxidant:
A mixture of Compound (C-a) (50.0 g), manganese (IV) oxide (152.8 g) and dichloromethane (500 mL) is stirred at about 20 °C. Upon completion of the reaction, Celite (15 g) was added. The resulting slurry was filtered through Celite (20 g) and the filter cake was rinsed with dichloromethane (500 mL). The filtrate was concentrated and solvent exchanged into cyclopentyl methyl ether (250 mL). The resulting solution was warmed to about 60 °C and treated with an aqueous potassium hydroxide solution (250 mL, 10wt%). The biphasic mixture is stirred at about 45 °C for about 12 hours. The phases are then separated and the organic phase is concentrated to a volume of about 150 mL. The concentrate is filtered, seeded with Compound (D-a) seed and agitated at about 40 °C to obtain a slurry. Methyl tert-butyl ether (450 mL) was added to the slurry over 30 minutes and the resulting mixture was cooled to about 20 °C. The precipitated solid was filtered, rinsed with methyl tert-butyl ether (250 mL) and dried in a vacuum oven at about 40 °C to obtain Compound (D-a).
Alternative Preparation of Compound (D-a) through catalytic dehydrogenation
A mixture of Compound (C-a) (2.5 g, 2.7 mmol, 1 equiv), 5% Pd/Al203 (2.5 g) and 1-propanol (25 mL, degassed) was stirred at reflux under inert environment for about 5.5 hours. The reaction mixture was then cooled to ambient temperature and filtered through Celite, and the residue rinsed with 1-propanol (2 x 5 mL) to obtain a solution of Compound (D-a).
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, in a reaction scheme employing stoichiometric oxidants, alternative oxidants may include manganese(IV) oxide, copper(II) acetate, copper(II) trifluoroacetate, copper(II) chloride, copper(II) bromide, bromine (Br2), iodine (I2), N-chlorosuccinimide, N-bromosuccinimide, N-iodosuccinimide, 1 ,4-benzoquinone, tetrachloro-l,4-benzoquinone (chloranil), eerie ammonium nitrate, hydrogen peroxide, tert-butyl hydroperoxide, άϊ-tert-butyl peroxide, benzoyl peroxide, oxygen ((¾), sodium hypochlorite, sodium hypobromite, tert-butyl hypochlorite, Oxone, diacetoxyiodobenzene, and bis(trifluoroacetoxy)iodobenzene. Various additives may be employed, and non-limiting examples may be carbonate bases (e.g., potassium carbonate, potassium bicarbonate, sodium carbonate, sodium bicarbonate, and the like), amines (e.g., triethylamine, diisopropylethylamine and the like), and acids (e.g., trifluoroacetic acid, trichloroacetic acid, benzoic acid, hydrochloric acid, sulfuric acid, phosphoric acid, ara-toluenesulfonic acid, methanesulfonic acid), sodium acetate, potassium acetate, and the like). The reaction temperature may range from about -10°C to 80 °C. The reaction may take place in solvents, such as halogenated solvents (e.g., dichloromethane, 1,2-dichloroethane, etc.), aromatic solvents (e.g., toluene, xylenes, etc.), ethereal solvents (tetrahydrofuran, 1,4-dioxane, cyclopentyl methyl ether, 1 ,2-dimethoxyethane, diglyme, triglyme, etc.), alcoholic solvents (e.g., methanol, ethanol, w-propanol, isopropanol, n-butanol, tert-butanol, tert-amyl alcohol, ethylene glycol, propylene glycol, etc.), ester solvents (e.g., ethyl acetate, isopropyl acetate, tert-butyl acetate, etc.), ketone solvents (e.g., acetone, 2-butanone, 4-methyl-2-pentanone, etc.), polar aprotic solvents (e.g., acetonitrile, Ν,Ν-dimethylformamide, N,N-dimethylacetamide, N-methyl-2-pyrrolidinone, pyridine, dimethyl sulfoxide, etc.), amine solvents (e.g., triethylamine, morpholine, etc.), acetic acid, and water.
In reaction schemes employing catalytic oxidants, alternative catalysts may include palladium catalysts (e.g., palladium(II) acetate, palladium(II) trifluoroacetate, palladium(II) chloride, palladium(II) bromide, palladium(II) iodide, palladium(II) benzoate, palladium(II) sulfate, tetrakis(triphenylphosphine)palladium(0), tris(dibenzylideneacetone)dipalladium(0), bis(tri-iert-butylphosphine)palladium(0), bis(triphenylphosphine)palladium(II) chloride, bis(acetonitrile)palladium(II) chloride, bis(benzonitrile)palladium(II) chloride, palladium on carbon, palladium on alumina, palladium on hydroxyapatite, palladium on calcium carbonate, palladium on barium sulfate, palladium(II) hydroxide on carbon), platinum catalysts (e.g., platinum on carbon, platinum(IV) oxide, chloroplatinic acid, potassium chloroplatinate), rhodium catalysts (e.g., rhodium on carbon, rhodium on alumina,
bis(styrene)bis(triphenylphosphine)rhodium(0)), ruthenium catalysts (e.g., ruthenium(II) salen, dichloro(para-cymene)ruthenium(II) dimer), iridium catalysts (e.g., iridium(III) chloride, (l,5-cyclooctadiene)diiridium(I) dichloride, bis(l,5-cyclooctadiene)iridium(I) tetrafluoroborate, bis(triphenylphosphine)(l,5-cyclooctadiene)iridium(I) carbonyl chloride, bis(triphenylphosphine)(l,5-cyclooctadiene)iridium(I) tetrafluoroborate), copper catalysts (e.g., copper(I) chloride, copper(II) chloride, copper(I) bromide, copper(II) bromide, copper(I) iodide, copper(II) iodide, copper(II) acetate, copper(II) trifluoroacetate, copper(I) trifluoromethanesulfonate, copper(II) trifluoromethanesulfonate, copper(II) sulfate), iron catalysts (e.g., iron(II) sulfate, iron(II) chloride, iron(III) chloride), vanadium catalysts (e.g., dichloro(ethoxy)oxovanadium, dichloro(isopropoxy)oxovanadium), manganese catalysts (e.g., manganese(rV) oxide, manganese(III) (salen) chloride), cobalt catalysts (e.g., cobalt(II) acetate, cobalt(II) chloride, cobalt(II) salen), indium(III) chloride, silver(I) oxide, sodium tungstate, quinone catalysts (e.g., 2,3-dichloro-5,6-dicyano-l,4-benzoquinone, 1,4-benzoquinone, and tetrachloro-l,4-benzoquinone (chloranil)).
Alternative co-oxidants can include, but are not limited to, sodium nitrite, copper(II) acetate, sodium persulfate, potassium persulfate, ammonium persulfate, sodium perborate, nitrobenzenesulfonate, 2,2,6,6-tetramethylpiperidine-l-oxyl (TEMPO), pyridine-N-oxide, hydrogen peroxide, tert-butyl hydroperoxide, di-tert-butyl peroxide, benzoyl peroxide, oxygen (02), sodium hypochlorite, sodium hypobromite, tert-butyl hypochlorite, oxone, diacetoxyiodobenzene, and bis(trifluoroacetoxy)iodobenzene.
Varoius hydrogen acceptors may be employed. Non-limiting examples can include unsaturated hydrocarbons (e.g., tert-butylethylene, tert-butyl acetylene, 2-hexyne, cyclohexene, and the like), acrylate esters (e.g., methyl acrylate, ethyl acrylate, isopropyl acrylate, tert-butyl acrylate, and the like), maleate esters (e.g., dimethyl maleate, diethyl maleate, diisopropyl maleate, dibutyl maleate, and the like), fumarate esters (e.g., dimethyl fumarate, diethyl fumarate, diisopropyl fumarate, dibutyl fumarate, and the like), and quinones (e.g. chloranil, 1 ,4-benzoquinone, etc.).
Alternative additives may be employed, such as carbonate bases (e.g., potassium carbonate, potassium bicarbonate, sodium carbonate, sodium bicarbonate, etc.), amine bases (e.g., triethylamine, diisopropylethylamine, etc.), phosphines (e.g., triphenylphosphine, tri(ort zotolyl)phosphine, tricyclohexylphosphine, tri-w-butylphosphine, tri-tert-butylphosphine, etc.), acids (e.g., trifluoroacetic acid, trichloroacetic acid, benzoic acid, hydrochloric acid, sulfuric acid, phosphoric acid, ara-toluenesulfonic acid, methanesulfonic acid, etc.), sodium acetate, N-hydroxyphthalimide, salen, 2,2 ‘-bipyri dine, 9,10-phenanthroline, and quinine.
The reaction can proceed at temperatures ranging from about 10 °C to about 120 °C. Various solvents can be employed, including but not limited to halogenated solvents (e.g., dichloromethane, 1,2-dichloroethane, and the like), aromatic solvents (e.g., toluene, xylenes, and the like), ethereal solvents (tetrahydrofuran, 1,4-dioxane, cyclopentyl methyl ether, 1,2-dimethoxyethane, diglyme, triglyme, and the like), alcoholic solvents (e.g., methanol, ethanol, w-propanol, isopropanol, w-butanol, tert-butanol, tert-amyl alcohol, ethylene glycol, propylene glyco, and the like), ester solvents (e.g., ethyl acetate, isopropyl acetate, tert-butyl acetate, and the like), ketone solvents (e.g., acetone, 2-butanone, 4-methyl-2-pentanone, and the like), polar aprotic solvents (e.g., acetonitrile, Ν,Ν-dimethylformamide, Ν,Ν-dimethylacetamide, N-methyl-2-pyrrolidinone, pyridine, dimethyl sulfoxide, and the like), amine solvents (e.g., triethylamine, morpholine, and the like), acetic acid, and water.

Acetyl chloride (135 mL, 5 equiv.) was added slowly to methanol (750 mL) under external cooling maintaining reaction temperature below 30 °C. The resulting methanolic hydrogen chloride solution was cooled to about 20 °C, and added slowly over about 1 hour to a solution of Compound (D-a) (300 g, 1 equiv.) in methanol (750 mL) held at about 60 °C, and rinsed forward with methanol (300 mL). The reaction mixture was agitated at about 60 °C until reaction was complete (about 1 hour), and then cooled to about 5 °C. The reaction mixture was adjusted to pH 7-8 by addition of sodium methoxide (25 wt. % solution in methanol, 370 mL) over about 20 minutes while maintaining reaction temperature below about 20 °C. Phosphoric acid (85 wt. %, 26 mL, 1 equiv.) and Celite (120 g) were added to the reaction mixture, which was then adjusted to about 20 °C, filtered, and the filter cake was rinsed with methanol (1050 mL). The combined filtrate was polish filtered and treated with phosphoric acid (85 wt. %, 104 mL, 4 equiv.). The mixture was was adjusted to about 60 °C, seeded with Compound (E-a) seed crystals (1.5 g), aged at about 60 °C for 4 hours and cooled slowly to about 20 °C over about 7.5 hours. The precipitated product was filtered, washed with methanol (2 x 600 mL), and dried in a vacuum oven at about 45 °C to provide
Compound (E-a). !H NMR (400 MHz, D20) δ 7.53-6.77 (comp m, 8H), 5.24-4.80 (comp m, 3H), 4.59-4.38 (comp m, 2H), 4.15-3.90 (m, 1H), 3.65-3.38 (comp m, 5H), 3.36-3.14 (comp m, 4H), 2.75 (s, 1H), 2.87-2.66 (m, 1H), 2.29-1.60 (comp m, 6H), 1.27 (d, 3H), 0.76 (m, 6H).
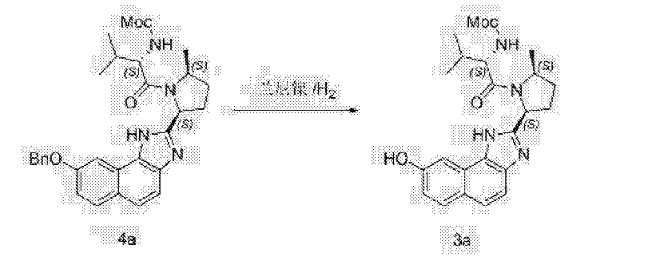
Alternative reagents and reaction conditions to those disclosed above may also be employed. Various deprotection agents are well known to those skilled in the art and include those disclosed in T.W. Greene & P.G.M. Wuts, Protective Groups in Organic Synthesis (4th edition) J. Wiley & Sons, 2007, hereby incorporated by reference in its entirety. For example, a wide range of acids may be used, including but not limited to phosphoric acid, trifluoroacetic acid, p-toluenesulfonic acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, 4-bromobenzenesulfonic acid, thionyl chloride,and trimethylsilyl chloride. A wide range of solvents may be employed, including but not limited to water, ethanol, acetonitrile, acetone, tetrahydrofuran, 1 ,4-dioxane, and toluene. Deprotection may proceed at temperatures ranging from about 20 °C to about 110 °C or from about 55 °C to about 65 °C.
A wide range of bases may be employed as a neutralization reagent. Non-limiting examples can include sodium phosphate dibasic, potassium phosphate dibasic, potassium bicarbonate, lithium hydroxide, sodium hydroxide, potassium hydroxide, triethylamine, N, N-diisopropylethylamine, and 4-methylmorpholine. Various solvents may be used for neutralization, such as water, ethanol, 1-propanol, 2-propanol, 1-butanol, 2-butanol, acetone, acetonitrile, 2-butanone, 4-methyl-2-pentanone, tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane, ethyl acetate, isopropyl acetate, dichloromethane, and dichloroethane.
Neutralization may proceed at temperatures ranging from about -20 °C to about 60 °C or about 5 °C to about 15 °C.
Various crystallization reagents can be employed. Non-limiting examples may be hydrochloric acid, hydrobromic acid, sulfuric acid, ethanesulfonic acid, benzenesulfonic acid, 4-bromobenzenesulfonic acid, oxalic acid, and glucuronic acid. Solvents for crystallization can include, but is not limited to, water, ethanol, 1-propanol, 2-propanol, and acetonitrile. Crystallization may proceed at temperatures ranging from about -20 °C to about 100 °C.
Free-Basing of Compound (E-a) to Prepare Compound (E)

ompound (E-a) OCH, H3CO- Compound (E)
Compound (E-a) (10.0 g, 10.1 mmol) was dissolved in water (100 g) and then dichloromethane (132 g) and 28% ammonium hydroxide (7.2 g) were added sequentially. The biphasic mixture was stirred for 45 minutes. Celite (2.2 g) was added, the mixture was filtered through a bed of additional Celite (5.1 g), and the phases were then separated. The lower organic phase was washed with water (50 g), filtered, and then concentrated by rotary evaporation to produce Compound (E). ‘H NMR (400 MHz, CD3OD) δ 8.35-7.17 (m, 8H), 5.6^1.68 (m, 3H), 4.41-3.96 (m, 2H), 3.96-3.72 (br s, 1H), 3.74-3.48 (m, 2H), 3.42 (d, 2H), 3.33 (s, 3H), 3.28 (s, 1H), 3.19-3.01 (m, 1H), 3.00-2.79 (m, 1H), 2.69-1.82 (m, 6H), 1.80-1.45 (m, 3H), 1.21-0.73 (m, 8H).
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, tris-hydrochloride salts of Compound (E) may be used. Various bases may be employed, such as sodium carbonate, potassium carbonate, sodium hydroxide, and potassium hydroxide. Various solvents, such as 2-methyltetrahydrofuran and ethyl acetate, may be employed. The temperature may range from about 15 °C to about 25 °C.
Alternative Free-Basing of Compound (E-b) to Prepare Compound (E)

Compound (E-b) (15.2 g) was dissolved in water (100 g) and then dichloromethane
(132 g) and 28% ammonium hydroxide (7.4 g) were added sequentially. The biphasic mixture was stirred for about 45 minutes. Celite (2.1 g) was added, the mixture was filtered through a bed of additional Celite (5.2 g), and the phases were then separated. The lower organic phase was washed with water (50 g), filtered, and then concentrated by rotary evaporation to produce Compound (E). *H NMR (400 MHz, CD3OD) δ 7.92-6.73 (m, 8H), 5.51-4.90 (m, 2H), 4.63-4.30 (m, 3H), 4.21-3.78 (m, 1H), 3.73-3.46 (m, 5H), 3.40-3.19 (m, 4H), 3.07-2.49 (m, 3H), 2.41-1.61 (m, 6H), 1.44-1.14 (m, 2H), 1.04-0.55 (m, 7H).
Salt Conversion of Compound (E-a) to Compound (E-b)

A solution of Compound (E-a) (10.0 g, 10.1 mmol), a solution of 37% HCI (10 g) in water (20 g), and acetonitrile (30 g)was warmed to about 50 °C and agitated for about lh. The solution was cooled to about 20 °C and acetonitrile (58 g) was charged to the reactor during which time a slurry formed. The slurry was stirred for about 21 h and then additional acetonitrile (39 g) was added. The slurry was cooled to about 0 °C, held for about 60 min and the solids were then isolated by filtration, rinsed with 7% (w/w) water in acetonitrile (22 g) previously cooled to about 5 °C. The wet cake was partially deliquored to afford
Compound (E-b). *H NMR (400 MHz, D20) δ 7.92-6.73 (m, 8H), 5.51^1.90 (m, 2H),
4.63-4.30 (m, 3H), 4.21-3.78 (m, 1H), 3.73-3.46 (m, 5H), 3.40-3.19 (m, 4H), 3.07-2.49 (m, 3H), 2.41-1.61 (m, 6H), 1.44-1.14 (m, 2H), 1.04-0.55 (m, 7H).

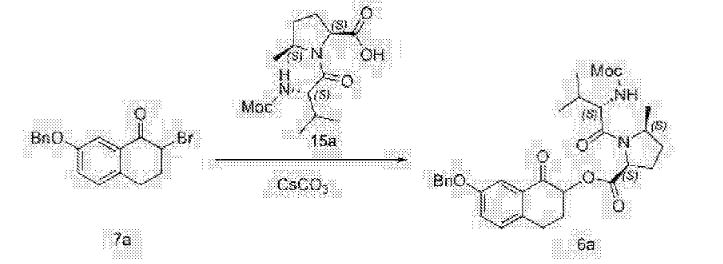
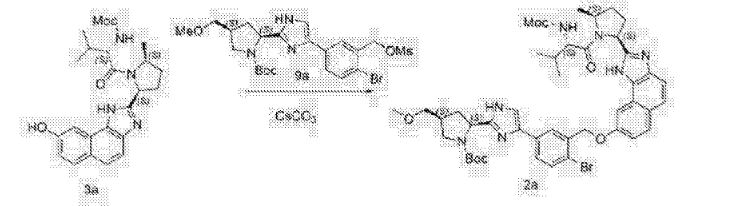
A flask was charged sequentially with 2-chloro-4,6-bis[3-(perfluorohexyl)propyloxy]-1,3,5-triazine (“CDMT”) (2.2 giv) and methanol (8.9 g) and the slurry was cooled to about 0 °C. To the mixture was added NMM (1.3 g) over about 5 minutes, maintaining an internal temperature of less than 20 °C. The solution was stirred for about 20 minutes to produce a solution of 4-(4,6-dimethoxy-l,3,5-triazin-2-yl)-4-methylmorpholinium chloride in methanol.
To a solution of Compound (E) (7.1 g) in dichloromethane (170 g) was added
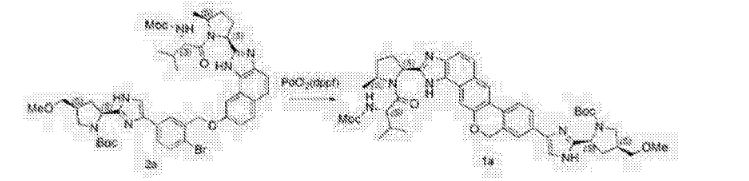
Compound (Γ) (2.8 g). The solution of 4-(4,6-dimethoxy-l,3,5-triazin-2-yl)-4-methylmorpholinium chloride in methanol was added over 2 minutes followed by a rinse of methanol (1.1 g). After about 2.5 h, the completed reaction solution was washed sequentially with aqueous 10% potassium bicarbonate solution (40 mL), 3% hydrochloric acid (40 mL), and aqueous 10% potassium bicarbonate solution (40 mL). The lower organic phase was washed with water (40 mL), filtered, and then concentrated by rotary evaporation to produce Compound (A). ¾ NMR (400 MHz, CD3OD) δ 8.56-6.67 (m, 13H), 5.76^1.94 (m, 4H), 4.86-4.67 (m, 1H), 4.47-3.98 (m, 1H), 3.98-2.72 (m, 15H), 2.74-1.77 (m, 7H), 1.77-1.40 (m, 2H), 1.39-0.53 (m, 8H).
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, tris-phosphate salts or tris-hydrochloride salts of Compound (G) may be used as alternative starting material. The reaction may take place at a temperature range of from about 10 °C to about 20 °C. Alternative coupling agents include, but are not limited to, EDC/HOBt, HATU, HBTU, TBTU, BOP, PyClOP, PyBOP, DCC/HOBt, COMU, EDCLOxyma, T3P, and 4-(4,6-dimethoxy-l,3,5-triazin-2-yl)-4-methylmorpholinium tetrafluoroborate. An alternative bases that may be employed can be diisopropylethylamine. The reaction may proceed in DMF and at temperatures ranging from about -20 °C to about 30 °C.
Salt Formation and Crystallization of Compound (A)
Crystallization of Compound (A-a)

A flask was charged with Compound (A) (10 g) and ethanol (125 mL) and was then warmed to about 45 °C. Concentrated hydrochloric acid (2.3 mL) was added followed by Compound (A-a) seed crystals (5 mg). The mixture was cooled to about 20 °C over about 5 h and held for about an additional 1 1 h. The solids were isolated by filtration, washed with ethanol (2 x 20 mL), and deliquored to produce Compound (A-a). !H NMR (400 MHz, CD3OD) δ 8.94-7.22 (m, 14H), 5.78-5.1 1 (m, 5H), 4.53-4.04 (m, 1H), 3.99-3.57 (m, 10H), 3.57-3.41 (m, 2H), 2.99-2.24 (m, 5H), 2.24-1.85 (m, 3H), 1.80-1.50 (m, 2H), 1.39-0.73 (m, 8H).
Alternative Crystallization of Compound (A-b)

A reaction vessel was charged with Compound (A) (25.0 g) followed by ethanol (125 mL) and 10% H3PO4 (250 mL). The solution was seeded with Compound (A-b) (100 mg) and stirred for about 17.5 h. The solids were isolated by filtration, washed with ethanol (2 x 5 mL), deliquored, and dried in a vacuum oven to produce Compound (A-b). JH NMR (400 MHz, D20) δ 7.76-6.48 (m, 13H), 5.53^1.90 (m, 3H), 4.60-4.32 (m, 2H), 4.29-3.76 (m, 1H), 3.70-2.75 (m, 14H), 2.66-1.51 (m, 8H), 1.51-1.09 (m, 3H), 1.05-0.45 (m, 7H).
Alternative reagents and reaction conditions to those disclosed above may also be employed. For example, alternative acids may be hydrochloric acid, hydrobromic acid, L-tartaric acid. Various solvents may be employed, such as methanol, ethanol, water, and isopropanol. The reaction may proceed at temperatures ranging from about 5 °C to about 60 °C.
Free-Basing of Compound (A)
Free-Basing of Compound (A-a) to Prepare Compound (A)

A reaction vessel was charged with Compound (A-a) (18.2 g) followed by ethyl acetate (188 g) and 10% potassium bicarbonate (188 g) and the mixture was stirred for about 25 minutes. The phases were separated and the upper organic phase was then washed with water (188 mL). The resulting organic solution was concentrated, ethanol (188 g) was added, and the solution was evaporated to produce a concentrate (75 g). The resulting concentrate added into water (376 g) to produce a slurry. The solids were isolated by filtration, washed with water (38 g), de liquored and dried in a vacuum oven at about 50 °C to produce
Compound (A).
Alternative Free-Basing of Compound (A-b) to Prepare Compound (A)

om poun –
A reaction vessel was charged with Compound (A-b) (3.0 g) followed by EtOAc (15 mL) and 10% KHCO3 (15 mL) and agitation was initiated. After about 5 h, the phases were separated and the organic phase was washed with water (15 mL) and then concentrated by rotary evaporation under vacuum. The residue was taken up in EtOH (4.5 mL) and then added to water (30 mL) to produce a slurry. After about 15 min, the solids were isolated by filtration rinsing forward water (3 x 3 mL). The solids were dried at about 50 to 60 °C vacuum oven for about 15 h to produce Compound (A).

PATENT
US 2015/0361085
https://patentscope.wipo.int/search/en/detail.jsf?docId=US153621930&redirectedID=true
An additional stable form screen was performed using the same procedure as described above but included a crystalline intermediate (Compound II shown below) as seeds. |

Compound II can be synthesized according to the methods described in WO 2013/075029 or U.S. Provisional Application No. 62/010,813. Needle-like particles were formed in butyronitrile, propionitrile, MEK/toluene, MEK/IPE and 2-pentanone/toluene. XRPD patterns of the wet solids were mostly consistent with each other with minor shifting in the peaks. The new form is named Compound I Form I, which is believed to be isostructural channel solvates with the respective solvents. After air drying all solids afforded amorphous XRPD patterns. |
Another stable form screen was performed using carbon (Darco G-60) treated Compound I, solvents, antisolvent (diisopropyl ether (IPE)), and seeds of Compound I Form I. This screen afforded crystalline solids from additional solvents as summarized in Table 1. The XRPD patterns of all of these solvates are consistent with Form I. The solvates were observed to convert to amorphous solids after drying. The XRPD patterns of Compound I were obtained in the experimental setting as follows: 45 kV, 40 mA, Kα1=1.5406 Å, scan range 2-40°, step size 0.0167°, counting time: 15.875 s. |
[TABLE-US-00002]
| TABLE 1 |
|
| Stable form screen of carbon treated Compound I |
| Solvents |
PLM |
Comments |
|
| Water |
Amorphous |
Slurry |
| Water/EtOH |
Amorphous |
Sticky phase coating |
| ACN/IPE |
Birefringent |
Slurry of needles |
| MeOH/IPE |
Solution |
Seeds dissolved |
| EtOH/IPE |
Solution |
Seeds dissolved |
| Acetone/IPE |
Birefringent |
Thick slurry of |
|
|
needles |
| IPA/IPE |
Amorphous |
Sticky coating |
| MEK/IPE |
Birefringent |
Thick slurry of |
|
|
needles |
| MIBK/IPE |
Birefringent |
White paste |
| DCM/IPE |
Birefringent |
Thick slurry of small |
|
|
needles |
| THF/IPE |
Solution |
Seeds dissolved |
| 2-MeTHF/IPE |
Amorphous |
slurry |
| EtOAc/IPE |
Birefringent |
Thick slurry of |
|
|
needles |
| IPAc/IPE |
Amorphous |
slurry |
| Toluene |
Amorphous |
Sticky coating |
|
|
The crystallinity of Compound I Form I can be improved by using a butyronitrile/butyl ether (BN/BE) mixture according to the following procedure. |
The crystallization experiment was started with 40 to 75 mg Compound I in 1.1 to 3.0 mL of a BN/BE in a ratio of 7:4 (anhydrous solvents). The sample was held at RT over P 2O 5 for 23 days without agitation, and crystals formed in the solution. Afterwards, the liquid phase was replaced with butyl ether and the solids were obtained by centrifuge. These solids, corresponding to Compound I Form I, were used for the subsequent step as seed. |
Purified Compound I (709.8 mg) was prepared from reflux of ethanol solution with Darco G-60 and was added to a new vial via a filter. While stirring, 7 mL of anhydrous butyronitrile (BN) was added. A clear orange solution was obtained. While stirring, 4 mL of anhydrous butyl ether (BE) was added slowly. To the solution was added 7.7 mg of Compound I Form I (from previous BN:BE crystallization experiment) as seed. The solution became cloudy and the seeds did not dissolve. The sample was stirred for ˜10 minutes before the agitation was stopped. The vial was capped and placed into a jar with some P 2O 5 solids at room temperature. After 6 days, a thin layer of bright yellow precipitate was observed on the wall and the bottom of the vial. The liquid phase was withdrawn and 3 mL of anhydrous butyl ether was added. Solids were scraped down with a spatula from the vial. The suspension was heated to about 30° C. for over half hour period and was held for ˜1 hour before cooling to 20° C. at about 0.1° C./min (without agitation). The sample was stored in ajar with P 2O 5 solids for 5 days. The sample was vacuum filtered using 0.22 μm nylon filter, washed with 2×200 μL of anhydrous butyl ether, and air dried under reduced pressure for about 5 minutes. |
XRPD analysis of the sample showed good very sharp peaks as shown in FIG. 1. The XRPD analysis setting was as follows: 45 kV, 40 mA, Kα1=1.5406 Å, scan range 1-40°, step size 0.0167°, counting time: 36.83 s. The characteristic peaks of crystalline Compound I Form I include: 2.9, 3.6, 4.8, 5.2, 6.0° 2θ ( FIG. 1). The XRPD pattern of Form I was successfully indexed, indicating that Form I is composed primarily of a single crystalline phase. Extremely large unit cell volume containing up to ˜60 API molecules in the unit cell was observed. The amorphous halo observed in the XRPD pattern could be a result of the size of the unit cell. Butyl ether stoichiometry could not be estimated. Two alternative indexing solutions were found: monoclinic and orthorhombic. |
DSC and TGA data confirmed that Form I is a solvated form. DSC shows a broad endotherm with onset at 109° C. and small endotherm with onset at 177° C. ( FIG. 2). TGA shows 22% weight loss below 150° C. ( FIG. 3). |
PATENT
CN 105294713
https://www.google.com/patents/CN105294713A?cl=en
https://patentimages.storage.googleapis.com/pdfs/2601c633c50937ffb780/CN105294713A.pdf
Example 12
Under nitrogen, was added l〇2g1 said, adding methylene burn 500 blood dissolved, 4mol / L fertilizer 1 1,4-dioxane SOOmL, football for 1 hour at room temperature, of the C (already burned: ethyl acetate 1: 1) point in the control board, the starting material spot disappeared, the reaction was stopped, the solvent was concentrated, was added (R & lt) -2- (methoxy several yl) -2-phenylacetic acid 29g, COMU60g, DMF blood 500, diisopropylethylamine 223M1,25 ° C reaction I h, ethyl acetate was added IL diluted, purified water is added IL painted twice, dried over anhydrous sulfate instrument, and concentrated, methanol was added SOOmL temperature 60 ° C dissolved, 250mL of purified water was slowly added dropwise, to precipitate a solid, the addition was completed, cooled to 50 ° C for 1 hour, cooled to room temperature, filtered, and concentrated to give Velpatasvir (GS-5816) product 90. 5g, 78. 2〇 yield / billion. H-NMR (400MHz, CDs isolated) 5 7. 94 – 7.67 (m, 4H), 7.59 of J = 9.1 Hz, 1H), 7. 52 (S, 1H), 7.48 – 7. 33 (m, 4H) , 7.11 of J = 18. 7Hz, 1H), 5.68 of J = 6.3Hz, 1H), 5.48 – 5.33 (m, 1H), 5.23 (dd, J = 24.1, 15.7Hz, 1H), 5.17 -5.03 (m, 3H), 4.22 (dd, J = 17.0, 9.6Hz, 1H), 4.16 – 4.01 (m, 1H), 3.91 (d, J = 24. 1 Hz, 1H), 3 83 -. 3. 68 (m, 1H), 3 68 -. 3. 59 (m, 3H), 3 59 -. 3. 49 (m, 3H), 3.38 (ddd, J = 15.9, 9.6, 5.7Hz, 2H), 3.28 – 3.14 (m, 5H), 3.10 (dd, J = 14.0, 8.2 Hz, 1H), 3.00 (dd, J = 17.8, 9.6Hz, 1H), 2.92 (dd, J = 14.5, 6.7 Hz, 1H), 2.73 – 2.41 (m, 2H), 2.40 – 2.11 (m, 2H), 2. 11 – 1.83 (m, 2H), 1.54 deduction J = 9. 7 Hz, 2H), 1.24 of J = 6.2Hz, 1H), 1.06 (t, J = 8.0 Hz, 1H), 0.99 of J = 6.8 Hz, 1H), 0. 94 (d, J = 6. 6Hz, 2H), 0. 85 (d, J = 6. 7Hz, 2H ).
Construction



Clip and foot notes
Velpatasvir only got its name last year and was previously known as GS-5816. That compound was only announced back in 2013 when Gilead showed the initial in vitrostudies on a handful of posters. [1] [2] Very little information is available on this follow-up compound. The following was pretty much the summary of their poster presentation.

To understand the medical significance of this study, Sofosbuvir is the best-in-class NS5B inhibitor from Gilead (see link for more information). [3] These inhibitors work the fastest when paired with a NS5A inhibitor like Daclatasvir or Ledipasvir (making up the Sofosbuvir+Ledipasvir = Harvoni combination) or the Viekira Pak combo. Disclosure: I am an employee of Bristol-Myers Squibb which produces Daclatasvir. However, HCV comprises of 7 different genotypes. Harvoni and Viekira Pak are approved against genotypes 1a, 1b. Harvoni is indicated for genotypes 4, 5, and 6. For the treatment of genotypes 2 and 3, sofosbuvir is generally combined with ribavirin or interferon which has notable side effects. While 70% of patients have genotype 1, for the remainder of patients with the other variants, they are still stuck with the more risky (and more expensive and longer) therapy.
I think this is the structure of GS-5816. It’s not yet published in any journal. [4]
For comparison, here is the structure of Ledipasvir, the first generation NS5A inhibitor used in Harvoni. Structurally speaking, they are pretty similar so it seems like GS-5816 is the product of good old fashioned medchem.
The clearest summary of the 4 Phase III trials can be found on Gilead’s website. [5]ASTRAL-1 was run on genotypes 1, 2, 4, 5, 6. [6] ASTRAL-2 focused on genotype 2. ASTRAL-3 focused on genotype 3. [7] ASTRAL-4 focused on HCV patients with Child-Pugh cirrhosis. [8] These patients previously had interferon treatment but had a poor response and are generally very sick.
I think that a few interesting things stand out. ASTRAL-1 occurred from July 2014 to December 2014 but upon a request from the FDA, ASTRAL-2 and 3 were started in September 2014-July 2015 in order to have an isolated study on genotypes 2 and 3. For a 24 week study that’s incredibly fast. As discussed elsewhere, clinical trials are often limited by the speed of patient enrollment and these studies can take years. [9] Here, they were able to find volunteers for a 1000 patient study within weeks. An interesting note about the clinical trial design, the ASTRAL-1 team knew that the historical cure rate was 85% and were able to correctly power the trial to get a statistically significant study on the first try. Also, deep sequencing was used to identify and stratify the HCV genotypes. In ASTRAL-1, 42% of the patients had NS5A resistance and 9% had NS5B resistance.
The market impact may be significant to Achillion which was a former partner of Gilead and a potential acquisition target. Achillion was working with Janssen on its own second generation NS5A inhibitor, odalasvir. This announcement may kill the market for a competing product as well as remove the acquisition hype.
How did Gilead come up with Velpatasvir? It really sounds like good solid science. Ledipasvir was developed to be a best-in-class NS5A inhibitor and it was recognized that it worked well with NS5B inhibitors. It was also understood that most of the NS5A inhibitors specific only towards certain N5SA genotypes and that there was a clear unmet need for patients with HCV genotypes 2 and 3. With the help of some computational modeling [10]Gilead developed assays for all of the HCV genotypes to screen for a pan-genotype NS5A inhibitor to follow up to their 2014 Ledipasvir trials and leveraging their strategic advantage in the HCV market, were able to quickly ramp up 4 major clinical trials to demonstrate the clinical efficacy of their next gen drug combination.
That’s really good science. Not long ago, Gilead stated that it was planning on eradicating HCV. This compound is a part of the Gilead license with Indian generic manufacturers but it seems like MSF is contesting that decision. [11] [12] With this drug Gilead is now another step closer towards that goal. [13]
Footnotes
[1] GS-5816, a Second-Generation HCV NS5A Inhibitor With Potent Antiviral Activity, Broad Genotypic Coverage, and a High Resistance Barrier
[2] Page on journal-of-hepatology.eu
[3] Christopher VanLang’s answer to How was Sovaldi (the drug now being marketed by Gilead), first discovered by Pharmasset?
[4] CAS # 1377049-84-7, Velpatasvir, GS 5816, Methyl [(2S)-1-[(2S,5S)-2-[9-[2-[(2S,4S)-1-[(2R)-2-[(methoxycarbonyl)amino]-2-phenylacetyl]-4-(methoxymethyl)pyrrolidin-2-yl]-1H-imidazol-5-yl]-1,11-dihydroisochromeno[4′,3′:6,7]naphtho[1,2-d]imidazol-2-yl]-5-methylpyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl]carbamate
[5] Page on gilead.com
[6] Sofosbuvir and Velpatasvir for HCV Genotype 1, 2, 4, 5, and 6 Infection — NEJM
[7] Sofosbuvir and Velpatasvir for HCV Genotype 2 and 3 Infection — NEJM
[8] Sofosbuvir and Velpatasvir for HCV in Patients with Decompensated Cirrhosis — NEJM
[9] Why do clinical trials for new drugs take several years? Remarkably, 72% of Americans are willing to be in them.
[10] Inhibition of hepatitis C virus NS5A by fluoro-olefin based γ-turn mimetics.
[11] Page on gilead.com
[12] MSF response to Gilead announcement on inclusion of hepatitis C drug GS-5816 in voluntary licence
[13] Gilead and Georgia to attempt Hep C eradication by Christopher VanLang on Making Drugs

SAVING LIVES
The Gilead team responsible for Harvoni: Front row, from left: John Link, Chris Yang, Rowchanak Pakdaman, Bob Scott, and Benjamin Graetz. Back row, from left: Erik Mogalian and Bruce Ross. Not pictured: Michael Sofia.
Credit: Gilead Sciences
Gilead’s Harvoni is a combination of two antiviral agents, sofosbuvir and ledipasvir. “In hepatitis C, the virus mutates so rapidly that to overcome resistance, we use a combination of drugs, and each one pulls their own weight in the process,” says John Link, who discovered ledipasvir.
Link says that the amount of interdisciplinary collaboration on the drug was unprecedented for the company. “Once ledipasvir was discovered, the process chemists were right there with us understanding the kinds of things we were doing, and medicinal chemists and process chemists worked on making material to scale for preclinical studies,” he says. “We all realized this was our moment to make a difference for patients with hepatitis C.”
Harvoni is the first once-a-day pill for treatment of chronic hepatitis C, and it has a cure rate in the U.S. of 94-99%. The drug is an alternative to injected interferon treatment, which has been associated with significant side effects.
“The high cure rates that we saw in our clinical trials are really amazing,” Link says. “Before we had these compounds, I had only hoped that we could equal something like interferon-type regimens in cure rates, without all the horrible side effects. To dramatically exceed them is important for patients.”
Harvoni patients can attest to the drug’s effectiveness. Mark Melancon, who had contracted hepatitis C 25 years ago, says that after taking Harvoni, he now has no trace of the virus in his body, and his liver is beginning to repair itself. “Four weeks into it, and the virus was gone. Not detectable,” he says. “To have this virus hanging over my head for 25 years and then it was just gone, I can’t explain the feeling. The people who worked hard on this medication, they need to know that I appreciate it.”

REFERENCES
https://www.eiseverywhere.com/file_uploads/c2a2b5664a374fe807c0b95bb546321d_JordanFeld.pdf
| WO2013075029A1 * |
Nov 16, 2012 |
May 23, 2013 |
Gilead Sciences, Inc. |
Condensed imidazolylimidazoles as antiviral compounds |
References
1: Kanda T. Interferon-free treatment for HCV-infected patients with decompensated cirrhosis. Hepatol Int. 2016 Jun 9. [Epub ahead of print] Review. PubMed PMID: 27282879.
2: Gane EJ, Schwabe C, Hyland RH, Yang Y, Svarovskaia E, Stamm LM, Brainard DM, McHutchison JG, Stedman CA. Efficacy of the Combination of Sofosbuvir, Velpatasvir, and the NS3/4A Protease Inhibitor GS-9857 in Treatment-naïve or Previously Treated Patients with HCV Genotype 1 or 3 Infections. Gastroenterology. 2016 May 27. pii: S0016-5085(16)34513-9. doi: 10.1053/j.gastro.2016.05.021. [Epub ahead of print] PubMed PMID: 27240903.
3: Schreiber J, McNally J, Chodavarapu K, Svarovskaia E, Moreno C. Treatment of a patient with genotype 7 HCV infection with sofosbuvir and velpatasvir. Hepatology. 2016 May 14. doi: 10.1002/hep.28636. [Epub ahead of print] PubMed PMID: 27177605.
4: Feld JJ, Zeuzem S. Sofosbuvir and Velpatasvir for Patients with HCV Infection. N Engl J Med. 2016 Apr 28;374(17):1688-9. PubMed PMID: 27135095.
5: Curry MP, Charlton M. Sofosbuvir and Velpatasvir for Patients with HCV Infection. N Engl J Med. 2016 Apr 28;374(17):1688. PubMed PMID: 27135094.
6: Assy N, Barhoum M. Sofosbuvir and Velpatasvir for Patients with HCV Infection. N Engl J Med. 2016 Apr 28;374(17):1687. doi: 10.1056/NEJMc1601160#SA1. PubMed PMID: 27119243.
7: Foster GR, Mangia A, Sulkowski M. Sofosbuvir and Velpatasvir for Patients with HCV Infection. N Engl J Med. 2016 Apr 28;374(17):1687-8. doi: 10.1056/NEJMc1601160. PubMed PMID: 27119242.
8: Smolders EJ, de Kanter CT, van Hoek B, Arends JE, Drenth JP, Burger DM. Pharmacokinetics, Efficacy, and Safety of Hepatitis C Virus Drugs in Patients with Liver and/or Renal Impairment. Drug Saf. 2016 Jul;39(7):589-611. doi: 10.1007/s40264-016-0420-2. Review. PubMed PMID: 27098247.
9: Majumdar A, Kitson MT, Roberts SK. Systematic review: current concepts and challenges for the direct-acting antiviral era in hepatitis C cirrhosis. Aliment Pharmacol Ther. 2016 Jun;43(12):1276-92. doi: 10.1111/apt.13633. Epub 2016 Apr 18. Review. PubMed PMID: 27087015.
10: Kahveci AS, Tahan V. Sofosbuvir and Velpatasvir: A complete pan-genotypic treatment for HCV patients. Turk J Gastroenterol. 2016 Mar;27(2):205-6. doi: 10.5152/tjg.2016.160000. PubMed PMID: 27015627.
11: Younossi ZM, Stepanova M, Feld J, Zeuzem S, Jacobson I, Agarwal K, Hezode C, Nader F, Henry L, Hunt S. Sofosbuvir/velpatasvir improves patient-reported outcomes in HCV patients: Results from ASTRAL-1 placebo-controlled trial. J Hepatol. 2016 Jul;65(1):33-9. doi: 10.1016/j.jhep.2016.02.042. Epub 2016 Mar 5. PubMed PMID: 26956698.
12: Gentile I, Scotto R, Zappulo E, Buonomo AR, Pinchera B, Borgia G. Investigational direct-acting antivirals in hepatitis C treatment: the latest drugs in clinical development. Expert Opin Investig Drugs. 2016 May;25(5):557-72. doi: 10.1517/13543784.2016.1161023. Epub 2016 Mar 21. PubMed PMID: 26934419.
13: Asselah T, Boyer N, Saadoun D, Martinot-Peignoux M, Marcellin P. Direct-acting antivirals for the treatment of hepatitis C virus infection: optimizing current IFN-free treatment and future perspectives. Liver Int. 2016 Jan;36 Suppl 1:47-57. doi: 10.1111/liv.13027. Review. PubMed PMID: 26725897.
14: Bourlière M, Adhoute X, Ansaldi C, Oules V, Benali S, Portal I, Castellani P, Halfon P. Sofosbuvir plus ledipasvir in combination for the treatment of hepatitis C infection. Expert Rev Gastroenterol Hepatol. 2015;9(12):1483-94. doi: 10.1586/17474124.2015.1111757. Epub 2015 Nov 23. PubMed PMID: 26595560.
15: Foster GR, Afdhal N, Roberts SK, Bräu N, Gane EJ, Pianko S, Lawitz E, Thompson A, Shiffman ML, Cooper C, Towner WJ, Conway B, Ruane P, Bourlière M, Asselah T, Berg T, Zeuzem S, Rosenberg W, Agarwal K, Stedman CA, Mo H, Dvory-Sobol H, Han L, Wang J, McNally J, Osinusi A, Brainard DM, McHutchison JG, Mazzotta F, Tran TT, Gordon SC, Patel K, Reau N, Mangia A, Sulkowski M; ASTRAL-2 Investigators; ASTRAL-3 Investigators. Sofosbuvir and Velpatasvir for HCV Genotype 2 and 3 Infection. N Engl J Med. 2015 Dec 31;373(27):2608-17. doi: 10.1056/NEJMoa1512612. Epub 2015 Nov 17. PubMed PMID: 26575258.
16: Feld JJ, Jacobson IM, Hézode C, Asselah T, Ruane PJ, Gruener N, Abergel A, Mangia A, Lai CL, Chan HL, Mazzotta F, Moreno C, Yoshida E, Shafran SD, Towner WJ, Tran TT, McNally J, Osinusi A, Svarovskaia E, Zhu Y, Brainard DM, McHutchison JG, Agarwal K, Zeuzem S; ASTRAL-1 Investigators. Sofosbuvir and Velpatasvir for HCV Genotype 1, 2, 4, 5, and 6 Infection. N Engl J Med. 2015 Dec 31;373(27):2599-607. doi: 10.1056/NEJMoa1512610. Epub 2015 Nov 16. PubMed PMID: 26571066.
17: Curry MP, O’Leary JG, Bzowej N, Muir AJ, Korenblat KM, Fenkel JM, Reddy KR, Lawitz E, Flamm SL, Schiano T, Teperman L, Fontana R, Schiff E, Fried M, Doehle B, An D, McNally J, Osinusi A, Brainard DM, McHutchison JG, Brown RS Jr, Charlton M; ASTRAL-4 Investigators. Sofosbuvir and Velpatasvir for HCV in Patients with Decompensated Cirrhosis. N Engl J Med. 2015 Dec 31;373(27):2618-28. doi: 10.1056/NEJMoa1512614. Epub 2015 Nov 16. PubMed PMID: 26569658.
18: Pianko S, Flamm SL, Shiffman ML, Kumar S, Strasser SI, Dore GJ, McNally J, Brainard DM, Han L, Doehle B, Mogalian E, McHutchison JG, Rabinovitz M, Towner WJ, Gane EJ, Stedman CA, Reddy KR, Roberts SK. Sofosbuvir Plus Velpatasvir Combination Therapy for Treatment-Experienced Patients With Genotype 1 or 3 Hepatitis C Virus Infection: A Randomized Trial. Ann Intern Med. 2015 Dec 1;163(11):809-17. doi: 10.7326/M15-1014. Epub 2015 Nov 10. PubMed PMID: 26551263.
19: Everson GT, Towner WJ, Davis MN, Wyles DL, Nahass RG, Thuluvath PJ, Etzkorn K, Hinestrosa F, Tong M, Rabinovitz M, McNally J, Brainard DM, Han L, Doehle B, McHutchison JG, Morgan T, Chung RT, Tran TT. Sofosbuvir With Velpatasvir in Treatment-Naive Noncirrhotic Patients With Genotype 1 to 6 Hepatitis C Virus Infection: A Randomized Trial. Ann Intern Med. 2015 Dec 1;163(11):818-26. doi: 10.7326/M15-1000. Epub 2015 Nov 10. PubMed PMID: 26551051.
20: Mogalian E, German P, Kearney BP, Yang CY, Brainard D, McNally J, Moorehead L, Mathias A. Use of Multiple Probes to Assess Transporter- and Cytochrome P450-Mediated Drug-Drug Interaction Potential of the Pangenotypic HCV NS5A Inhibitor Velpatasvir. Clin Pharmacokinet. 2016 May;55(5):605-13. doi: 10.1007/s40262-015-0334-7. PubMed PMID: 26519191.

| Patent ID |
Date |
Patent Title |
| US2015064252 |
2015-03-05 |
SOLID DISPERSION FORMULATION OF AN ANTIVIRAL COMPOUND |
| US2015064253 |
2015-03-05 |
COMBINATION FORMULATION OF TWO ANTIVIRAL COMPOUNDS |
| US8940718 |
2015-01-27 |
Antiviral compounds |
| US8921341 |
2014-12-30 |
Antiviral compounds |
| US2014357595 |
2014-12-04 |
METHODS OF PREVENTING AND TREATING RECURRENCE OF A HEPATITIS C VIRUS INFECTION IN A SUBJECT AFTER THE SUBJECT HAS RECEIVED A LIVER TRANSPLANT |
| US2014343008 |
2014-11-20 |
HEPATITIS C TREATMENT |
| US2014316144 |
2014-10-23 |
ANTIVIRAL COMPOUNDS |
| US2014309432 |
2014-10-16 |
ANTIVIRAL COMPOUNDS |
| US2014212491 |
2014-07-31 |
COMBINATION FORMULATION OF TWO ANTIVIRAL COMPOUNDS |
| US2014018313 |
2014-01-16 |
ANTIVIRAL COMPOUNDS |
Velpatasvir
 |
| Systematic (IUPAC) name |
|
(2S)-2-{[hydroxy(methoxy)methylidene]amino}-1-[(2S,5S)-2-(17-{2-[(2S,4S)-1-[(2R)-2-{[hydroxy(methoxy)methylidene]amino}-2-phenylacetyl]-4-(methoxymethyl)pyrrolidin-2-yl]-1H-imidazol-5-yl}-21-oxa-5,7-diazapentacyclo[11.8.0.0³,¹¹.0⁴,⁸.0¹⁴,¹⁹]henicosa-1(13),2,4(8),6,9,11,14(19),15,17-nonaen-6-yl)-5-methylpyrrolidin-1-yl]-3-methylbutan-1-one
|
| Identifiers |
| CAS Number |
1377049-84-7 |
| PubChem |
CID 67683363 |
| ChemSpider |
34501056 |
| UNII |
KCU0C7RS7Z  |
| Chemical data |
| Formula |
C49H54N8O8 |
| Molar mass |
883.02 g·mol−1 |
//////////////VELPATASVIR, GS-5816, GILEAD SCIENCES, Epclusa , FDA 2016, велпатасвир,فالباتاسفير , 维帕他韦 , велпатасвир, فالباتاسفير , 维帕他韦 , Elizabeth Bacon, Sheila Zipfel
C[C@H]1CC[C@H](N1C(=O)[C@H](C(C)C)NC(=O)OC)C2=NC3=C(N2)C=CC4=CC5=C(C=C43)OCC6=C5C=CC(=C6)C7=CN=C(N7)[C@@H]8C[C@@H](CN8C(=O)[C@@H](C9=CC=CC=C9)NC(=O)OC)COC

/////



































 .
.










 Open Access
Open Access