
AMG-900
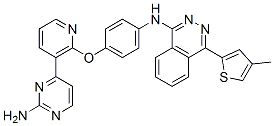
N-(4-((3-(2-aminopyrimidin-4-yl)pyridin-2-yl)oxy)phenyl)-4-(4-methylthiophen-2-yl)phthalazin-1-amine.
N-(4-(3-(2-Aminopyrimidin-4-yl)pyridin-2-yloxy)phenyl)-4-(4-methylthiophen-2-yl)phthalazin-1-amine
Phase I
Amgen Inc. INNOVATOR
| Inventors |
Victor J. Cee, Holly L. Deak, Bingfan Du,Stephanie D. Geuns-Meyer, Brian L. Hodous,Hanh Nho Nguyen, Philip R. Olivieri, Vinod F. Patel, Karina Romero, Laurie Schenkel,Less « |
| Applicant |
Amgen Inc. |
An aurora kinase (ARK) inhibitor potentially for the treatment of leukemia and solid tumours.
CAS No. 945595-80-2
In 2014, orphan drug designation was assigned in the U.S. for the treatment of ovarian cancer
| Molecular Formula: |
C28H21N7OS |
| Molecular Weight: |
503.57764 g/mo |
|
AMG 900; AMG-900; 945595-80-2; AMG900; UNII-9R2G075611; N-(4-((3-(2-aminopyrimidin-4-yl)pyridin-2-yl)oxy)phenyl)-4-(4-methylthiophen-2-yl)phthalazin-1-amine; |
AMG 900 is a small-molecule inhibitor of Aurora kinases A, B and C with potential antineoplastic activity. Aurora kinase inhibitor AMG 900 selectively binds to and inhibits the activities of Aurora kinases A, B and C, which may result in inhibition of cellular division and proliferation in tumor cells that overexpress these kinases. Aurora kinases are serine-threonine kinases that play essential roles in mitotic checkpoint control during mitosis and are overexpressed by a wide variety of cancer cell types. Check for active clinical trials or closed clinical trials using this agent

AMG 900 is a potent and highly selective pan-Aurora kinases inhibitor for Aurora A/B/C with IC50 of 5 nM/4 nM /1 nM; >10-fold selective for Aurora kinases than p38α, Tyk2, JNK2, Met and Tie2.
IC50 Value: 5 nM(Aurora A); 4 nM(Aurora B); 1 nM(Aurora C)
Target: pan-Aurora
in vitro: AMG 900 is a novel class of ATP-competitive phthalazinamine small molecule inhibitors of aurora kinases. In HeLa cells, AMG 900 inhibits autophosphorylation of aurora-A and -B as well as phosphorylation of histone H3 on Ser, a proximal substrate of aurora-B. The predominant cellular response of tumor cells to AMG 900 treatment is aborted cell division without a prolonged mitotic arrest, which ultimately results in cell death. AMG 900 inhibits the proliferation of 26 tumor cell lines, including cell lines resistant to the antimitotic drug paclitaxel and to other aurora kinase inhibitors (AZD1152, MK-0457, and PHA-739358), at low nanomolar concentrations (about 2- 3 nM). Furthermore, AMG 900 is active in an AZD1152-resistant HCT116 variant cell line that harbors an aurora-B mutation (W221L) [1].
in vivo: Oral administration of AMG 900 blocks the phosphorylation of histone H3 in a dose-dependent manner and significantly inhibited the growth of HCT116 tumor xenografts. AMG 900 is broadly active in multiple xenograft models, including 3 multidrugresistant xenograft models, representing 5 tumor types [1]. AMG 900 exhibits a low-to-moderate clearance and a small volume of distribution. Its terminal elimination half-life ranged from 0.6 to 2.4 hours. AMG 900 is well-absorbed in fasted animals with an oral bioavailability of 31% to 107%. Food intake has an effect on rate (rats) or extent (dogs) of AMG 900 oral absorption. The clearance and volume of distribution at steady state in humans are predicted to be 27.3 mL/h/kg and 93.9 mL/kg, respectively. AMG 900 exhibits acceptable PK properties in preclinical species and is predicted to have low clearance in humans [2].
In mammalian cells, the aurora kinases (aurora-A, -B, and -C) play essential roles in regulating cell division. The expression of aurora-A and -B is elevated in a variety of human cancers and is associated with high proliferation rates and poor prognosis, making them attractive targets for anticancer therapy. AMG 900 is an orally bioavailable, potent, and highly selective pan-aurora kinase inhibitor that is active in taxane-resistant tumor cell lines. In tumor cells, AMG 900 inhibited autophosphorylation of aurora-A and -B as well as phosphorylation of histone H3 on Ser(10), a proximal substrate of aurora-B. The predominant cellular response of tumor cells to AMG 900 treatment was aborted cell division without a prolonged mitotic arrest, which ultimately resulted in cell death. AMG 900 inhibited the proliferation of 26 tumor cell lines, including cell lines resistant to the antimitotic drug paclitaxel and to other aurora kinase inhibitors (AZD1152, MK-0457, and PHA-739358), at low nanomolar concentrations. Furthermore, AMG 900 was active in an AZD1152-resistant HCT116 variant cell line that harbors an aurora-B mutation (W221L). Oral administration of AMG 900 blocked the phosphorylation of histone H3 in a dose-dependent manner and significantly inhibited the growth of HCT116 tumor xenografts. Importantly, AMG 900 was broadly active in multiple xenograft models, including 3 multidrug-resistant xenograft models, representing 5 tumor types. AMG 900 has entered clinical evaluation in adult patients with advanced cancers and has the potential to treat tumors refractory to anticancer drugs such as the taxanes.

MG 900 is an orally bioavailable, potent, and highly selective pan-aurora kinase inhibitor that is active in taxane-resistant tumor cell lines. In tumor cells, AMG 900 inhibited autophosphorylation of aurora-A and -B as well as phosphorylation of histone H3 on Ser10, a proximal substrate of aurora-B. The predominant cellular response of tumor cells to AMG 900 treatment was aborted cell division without a prolonged mitotic arrest, which ultimately resulted in cell death. AMG 900 inhibited the proliferation of 26 tumor cell lines, including cell lines resistant to the antimitotic drug paclitaxel and to other aurora kinase inhibitors (AZD1152, MK-0457, and PHA-739358), at low nanomolar concentrations. Furthermore, AMG 900 was active in an AZD1152-resistant HCT116 variant cell line that harbors an aurora-B mutation (W221L). Oral administration of AMG 900 blocked the phosphorylation of histone H3 in a dose-dependent manner and significantly inhibited the growth of HCT116 tumor xenografts. Importantly, AMG 900 was broadly active in multiple xenograft models, including 3 multidrug-resistant xenograft models, representing 5 tumor types. AMG 900 has entered clinical evaluation in adult patients with advanced cancers and has the potential to treat tumors refractory to anticancer drugs such as the taxanes. (Source: Cancer Res; 70(23); 9846–54.)
Clinical Information of AMG 900
| Product Name |
Sponsor Only |
Condition |
Start Date |
End Date |
Phase |
Last Change Date |
| AMG 900 |
Amgen Inc |
Leukemia |
31-JUL-11 |
31-JUL-14 |
Phase 1 |
14-SEP-13 |
| Amgen Inc |
Advanced solid tumor |
30-APR-09 |
30-JUN-13 |
Phase 1 |
10-SEP-13 |


PATENT
WO 2007087276
http://www.google.co.in/patents/WO2007087276A1?cl=en
PATENT
WO 2015084649
https://google.com/patents/WO2015084649A1?cl=en
The compound, N-(4-((3-(2-amino-4-pyrimidinyl)-2-pyridinyl)oxy)phenyl)- 4-(4-methyl-2-thienyl)-l-phthalazinamine, also chemically named as 4-((3-(2-amino- pyrimidin-4-yl)-pyridin-2-yl)oxy)phenyl-(4-(4-methyl-thiophen-2-yl)-phthalazin-l- yl)amine, and is referred to herein as “AMG 900” has a chemical structure of

AMG 900 is an ATP competitive small molecule Aurora kinase inhibitor that is highly potent and selective for Aurora kinases A, B and C. AMG 900 is disclosed in US patent publication no. 20070185111, which published on August 9, 2007 and issued as U.S. Patent No. 7,560,551. AMG 900 is further disclosed in US patent publication no.
20090163501, now US patent no 8,022,221. Various uses and applications of AMG 900 are described in patent publications US20120028917 and WO2013149026. AMG 900 is being clinically evaluated primarily for its safety, tolerability and pharmacokinetic (PK) profile in human phase I trials for (1) advanced solid tumors (US Clinical Trial Id No. NCT00858377), and (2) for acute leukemias (US Clinical Trial Id No. NCT1380756).
Different solid state forms of a given compound are typically investigated to determine whether or not a particular form possesses and/or exhibits desirable properties allowing that compound to be clinically and/or commercially developed. Such beneficial and advantageous properties, by way of example, include without limitation, crystallinity, improved thermodynamic stability, non-hygroscopicity, high purity, minimal to total absence of moisture and/or residual solvents, chemical stability, high yielding synthetic process and/or manufacturability and reproducibility, desirable biopharmaceutical properties including improved dissolution characteristics and increased bioavailability, absence or reduced toxicities due to reduced or limited exposure, rate of exposure or release, or related to counter ions, good bulk and formulation properties including good flow, bulk density, desirable particle size and the like, or a combination of the aforementioned characteristic attributes.
Generally when a compound, also referred to herein as drug substance (DS), has been identified as a developmental candidate, the DS is screened to identify potentially beneficial polymorphic, crystalline or solid state forms of the compound and/or a pharmaceutically acceptable salt thereof. X-ray diffraction, Raman, solid state NMR and a melting point temperature and/or a melting point temperature range have been typically used to monitor or screen and identify the different polymorphic form of the DS.
Different polymorphic forms of a given DS can have an impact on that compound’s solubility, stability and bioavailability. Also, it is important to monitor possible changes in polymorphic forms of the DS during stability studies.
AMG 900 was previously isolated and identified as a free base compound. This compound exhibited rather lack-luster pharmacokinetic (PK) and/or pharmacodynamic (PD) properties, including poor aqueous solubility, poor bioavailability, poor absorption, poor target exposure and overall, a not-so-attractive in-vivo efficacy profile. Thus, there is a need to address and solve the technical problem of identifying alternative forms of AMG 900 to achieve substantially the same effect or an improved effect, including improved PK and PD profiles, as that of AMG 900 known in the art.
Example 1

Synthesis of N-(4-((3-(2-amino-4-pyrimidinylN)-2-pyridinylN)oxyN)phenylN)-4-(4-methyl-2-thienvD-l-phthalazinamine (AMG 900)
Step 1 : 4-(2-chloropyridin-3-yl)pyrimidin-2 -amine
In an argon purged 500 mL round bottom flask placed in an isopropanol bath, was added sodium metal (3.40g, 148mmol) slowly to methanol (180mL). The mixture was stirred at room temperature (RT) for about 30 minutes. To this was added guanidine hydrochloride (12.0 mL, 182 mmol) and the mixture was stirred at RT for 30 minutes, followed by addition of (E)-l-(2-chloropyridin-3-yl)-3-(dimethylamino)prop-2-en-l-one (12.0 g, 57.0 mmol), attached air condenser, moved reaction to an oil bath, where it was heated to about 50 °C for 24 hr. Approximately half of the methanol was evaporated under reduced pressure and the solids were filtered under vacuum, then washed with saturated sodium bicarbonate (NaHCO and H^O, air dried to yield 4-(2-chloropyridin-3-yl)pyrimidin-2-amine as off white solid. MS m/z = 207 [M+l]+. Calc’d for C9H7C1N4: 206.63.
Step 2: 4-(2-(4-aminophenoxy)pyridin-3-yl)pyrimidin-2-amine
To a resealable tube was added 4-aminophenol (1.3 g, 12 mmol), cesium carbonate (7.8 g, 24 mmol), and DMSO (16 ml, 0.75 M). The mixture was heated to 100 °C for 5 minutes, and then 4-(2-chloropyridin-3-yl)pyrimidin-2 -amine (2.5 g, 12 mmol) was added, and the reaction mixture was heated to 130 °C overnight. Upon completion, as judged by LCMS, the reaction mixture was allowed to cool to RT and diluted with water. The resulting precipitate was filtered, and the solid washed with water and diethyl ether. The solid was then taken up in 9: 1 CH2Cl2:MeOH and passed through a pad of silica gel with 9:1 CH2Cl2:MeOH as eluent. The solvent was concentrated in vacuo to provide the desired product, 4-(2-(4-aminophenoxy)pyridin-3-yl)pyrimidin-2-amine. MS m/z = 280
[M+l]+. Calc’d for Ci5H13N50: 279.30.
Step 3: l-Chloro-4-(4-methylthiophen-2-yl)phthalazine
1 ,4-Dichlorophthalazine (1.40 g, 7.03 mmol), 4-methyltmophen-2-ylboronic acid (999 mg, 7.03 mmol), and PdCl2(DPPF) (721 mg, 985 μιηοΐ) were added into a sealed tube. The tube was purged with Argon. Then sodium carbonate (2.0 M in water) (7.74 ml, 15.5 mmol) and 1,4-dioxane (35.2 ml, 7.03 mmol) were added. The tube was sealed, stirred at RT for 5 min, and placed in a preheated oil bath at 110 °C. After 1 hr, LC-MS showed product and byproduct (double coupling), and starting material
dichlorophthalazme. The reaction was cooled to RT, filtered through a pad of celite with an aid of ethyl acetate (EtOAc), concentrated, and loaded onto column. The product was purified by column chromatography using Hex to remove the top spot, then 80:20 hexanes:EtOAc to collect the product. The product, 1 -chloro-4-(4-methylthiophen-2-yl)phthalazine was obtained as yellow solid. LC-MS showed that the product was contaminated with a small amount of dichlorophthalazme and biscoupling byproduct. MS m/z = 261 [M+l]+. Calcd for Ci3H9ClN2S: 260.12.
Step 4: N-(4-((3-(2-amino-4-pyrimidinyl)-2-pyridinyl)oxy)phenyl)-4-(4-methyl-2-thienyl)- 1 -phthalazinamine
To 4-(2-(4-aminophenoxy)pyridin-3-yl)pyrimidin-2 -amine and l-chloro-4-(4-methyl-2-thienyl)phthalazine was added tBuOH. The resulting mixture was heated at 100 °C in a sealed tube for 16 hours. The rection was diluted with diethyl ether and saturated sodium carbonate and vigorously shaken. The resulting solids were filtered and washed with water, diethyl ether and air dried to yield N-(4-((3-(2-amino-4-pyrimidinyl)-2-pyridinyl)oxy)phenyl)-4-(4-methyl-2-thienyl)-l -phthalazinamine as an off-white solid. MS m/z = 504 [M+H]+. Calc’d for C28 H21 N7 O S: 503.58.
Example 2

Alternative Synthesis of N-(4-((3-(2-amino-4-pyrimidinylN)-2-pyridinylN)oxyN)phenylN)-4-(4-methyl-2-thienvD-l-phthalazinamine (AMG 900)
Step 1 : 4-(2-chloropyridin-3-yl)pyrimidin-2 -amine
In an argon purged 500 mL round bottom flask placed in an isopropanol bath, was added sodium metal (3.40g, 148mmol) slowly to methanol (180mL). The mixture was stirred at room temperature (RT) for about 30 minutes. To this was added guanidine hydrochloride (12.0 mL, 182 mmol) and the mixture was stirred at RT for 30 minutes, followed by addition of (E)-l-(2-chloropyridin-3-yl)-3-(dimethylamino)prop-2-en-l-one (12.0 g, 57.0 mmol), attached air condenser, moved reaction to an oil bath, where it was heated to about 50 °C for 24 hr. Approximately half of the methanol was evaporated under reduced pressure and the solids were filtered under vacuum, then washed with saturated sodium bicarbonate (NaHCO and H^O, air dried to yield 4-(2-chloropyridin-3-yl)pyrimidin-2-amine as off white solid. MS m/z = 207 [M+l]+. Calc’d for C9H7C1N4: 206.63.
Step 2: 4-(2-(4-aminophenoxy)pyridin-3-yl)pyrimidin-2-amine
To a reaction vessel at ambient temperature was added 4-aminophenol (571 g, 5.25 mol, 1.05 equiv) followed by 4-(2-chloropyridin-3-yl)pyrimidin-2-amine (1064g, 97 wt%, 5.00 mol, 1.0 equiv) and DMSO (7110 ml, 7820 g, 7x the volume of 4-(2-chloropyridin-3-yl)pyrimidin-2 -amine). The reaction mixture was agitated and sparged with nitrogen gas for at least 10 minutes. Then a 50 weight % aqueous KOH solution (593 g, 5.25 mol, 1.05 equiv.) was added to the mixture while keeping the reaction
mixture temperature below about 40°C. The mixture was sparged with nitrogen gas for more than 5 minutes, the sparging tube was removed, and the reaction mixture was heated to 110 +/- 10 °C for at least 1.5 hrs. Upon completion, as judged by HPLC, the reaction mixture was allowed to cool to RT and diluted with 6N HC1 (42 mL, 0.25 mol, 0.05 equiv). The desired product, 4-(2-(4-aminophenoxy)pyridin-3-yl)pyrimidin-2 -amine was not isolated. Rather, it was formed in-situ and combined with the product of step 3 below, in step 4 to form the desired product.
Step 3: l-Chloro-4-(4-methylthiophen-2-yl)phthalazine
A separate reaction vessel was fitted with a reflux condenser and an addition funnel, and 4-(4-methylthiophen-2-yl)phthalazin-l(2H)-one (1,537 mg, 6.34 mol, 1.0 equivalent) was added to the reaction vessel. Acetonitrile (7540 mL, 5859 g, 5 V), was added and the reaction vessel was agitated to allow the starting material to dissolve. The vessel was then charged with phosphorus oxychloride (709 ml, 1166 g, 7.44 mol, 1.2 equivalents) and the reaction was heated to about 75 +/- 5 °C for a least 4 hrs. The reaction was cooled to about about 25 +/- 5 °C and held there for more than 24 hrs. N,N-diisopropylethylamine (3046 g, 4100 mL, 3.8 equivalents) was added to the reaction vessel and the temperature was maintained at <30°C. Pyridine (97g, 1.24 mol, 0.2 equiv) was added in a single portion followed by water (4100 g, 2.7V) over more than 30 minutes. The reaction mixture was stirred at ambient temperature ofr about 24 hrs. the mixture was filtered through a <25uM polypropylene filter and the rsulting mother liquor was diluted with 1 : 1 ACN:water (9000 mL total) and stirred for a minimum of 2 minutes. Filter off product solids as they precipitate. Collect mother liquor and washes to obtain additional product. Dry the filter cake, and additional product crops, under a constant stream of nitrogen gas for at least 14 hrs. Unlike the previous method, the present method avoids contamination of impurities, such as dichlorophthalazine and biscoupling byproduct, as seen via LC-MS. Yield: 1537 g (97.2 weight %). MS m/z = 261 [M+l]+. Calcd for Ci3H9ClN2S: 260.12.
Step 4: N-(4-((3-(2-amino-4-pyrimidinyl)-2-pyridinyl)oxy)phenyl)-4-(4-methyl-2-thienyl)- 1 -phthalazinamine
To the reaction mixture was added l-chloro-4(4-methylthiophen-2-yl)phthalazine
(1450g, 97.2 wt%, 5.40 mol, 1.08 equiv) rinding the addition port with DMSO (520 ml, 572 g, 0.5x the volume of 4-(2-chloropyridin-3-yl)pyrimidin-2-amine). The reaction mixture was again agitated and sparged with nitrogen gas for at least 10 minutes. The sparging tube was removed, and the reaction mixture was heated to 80 +/- 20 °C for at least 2 hrs. Upon completion, as judged by HPLC, the reaction mixture was allowed to cool to RT and N,N-diisopropylethylamine (776 g, 1045 mL, 6.0 mol, 1.2 equiv) was added and the mixture was kept at about 80 +/- 10°C. Filter the mixture at about 80oC into a separate reactor vessel rinsing with DMSO (1030 mL, 1133 g, 1 V). Then adjust the raction mixture temperature to about 70+/-5 °C and add 2-propanol (13200 mL, 10360 g, 12.75 V) over more than 15 minutes at about 70°C. As the reaction mistreu cools, the product begins to precipitate out of solution. Add more 2-propanol (8780 mL, 6900 g, 8.5V) to the solution slowly over more then 60 minutes at about 70°C. The reactor vessel was cooled to about 20°C over more than 60 minutes and let sit for over 2 hrs. The product was filtered through an Aurora filter with a >25uM polypropylene filter cloth. Additional crops were obtained from the mother liquors by diluting with additional 2-propanol. The filter cakes were dried under a constant stream of nitrogen gas for at least 14 hrs to provide the desired product, N-(4-((3-(2-amino-4-pyrimidinyl)-2-pyridinyl)oxy)phenyl)-4-(4-methyl-2-thienyl)-l-phthalazinamine as an off-white solid. Yield: 2831 g (88.8%); purity 99.7%. MS m/z = 504 [M+H]+. Calc’d for C28 H21 N7 O S: 503.58.
The starting material 1 used/shown in Example 2 was prepared as follows:

and starting material 3, thienyl substituted phthalazinone, shown in Example 2 was prepared as follows:

Starting material 3
Synthesis of 4-(5-methylthiophen-2-yl)phthalazin-l(2//)-one
Step 1 : 2-(Dimethylamino)isoindoline-1.3-dione
A solution of isobenzofuran-l,3-dione (5.00 g, 34 mmol) and N,N-dimethylhydrazine (2.9 ml, 37 mmol) in toluene (75 ml, 34 mmol) was added p-TsOH H20 (0.32 g, 1.7 mmol). The Dean-Stark apparatus and a condenser were attached. The mixture was refluxed. After 4 hr, LCMS showed mainly product. The reaction was cooled to rt. Toluene was removed under reduced pressure the crude was dissolved in DCM, washed with sat NaHC03, water, and brine. The organic was dried over MgS04, filtered, and concentrated. Light yellow solid was obtained. !H NMR showed mainly product, 2-(dimethylamino)isoindoline-l,3-dione. MS Calcd for C10H10N2O2: [M]+ = 190. Found: [M+H]+ = 191.
Step 2 : 2-(Dimethylamino)-3 -hydroxy-3 -(5 -methylthiophen-2 -vDisoindolin- 1 -one
A solution of 2-bromo-5-methylthiophene (0.60 mL, 5.3 mmol) in THF (11 mL) was purged with nitrogen and cooled to -78 °C. «-Butyllithium (2.2 mL, 5.5 mmol; 2.5 M in THF) was added and the mixture was stirred under nitrogen for 30 min. This solution was cannulated into a flask containing a solution of 2-(dimethylamino)isoindoline-l,3-dione (1.5 g, 7.9 mmol) in THF (16 mL) at -78 °C under nitrogen. The reaction was allowed to warm to -30 °C over an hour, at which point LCMS showed complete conversion of 2-bromo-5-methylthiophene to product. The reaction was quenched by careful addition of saturated aqueous NH4C1. The reaction mixture was diluted with dichloromethane and water, and the layers were separated. The aqueous portion was extracted with additional dichloromethane, and the combined organic layers were dried with MgS04, filtered, concentrated, and purified by silica gel chromatography eluting with 0-2% MeOH in dichloromethane to provide intermediate A, as a light yellow solid, 2-(dimethylamino)-3-hydroxy-3-(5-methylthiophen-2-yl)isoindolin-l-one (1.2 g, 80% yield). !H NMR (400 MHz, DMSO-4) δ 7.68-7.65 (m, 1H). 7.63-7.59 (m, 1H), 7.57-7.51 (m, 1H), 7.37 (d, 1H, J=8), 7.09 (s, 1H), 6.69-6.66 (m, 1H), 6.65-6.62 (m, 1H), 2.81 (s, 6H), 2.40 (s, 3H). 13C NMR (400 MHz, DMSO-de) δ 165.0, 147.3, 141.6, 139.3, 132.7, 129.49, 129.46, 125.0, 124.7, 123.0, 122.1, 88.4, 44.7, 14.9. FT-IR (thin film, cm ) 3347, 3215, 1673. MS Calcd for Ci2H7ClN2S: [M]+ = 288. Found: [M+H]+= 289.
HRMS Calcd for Ci5H16N202S: [M+H]+= 288.1005, [M+Na]+ = 311.0825. Found:
[M+H]+ = 289.1022, [M+Na]+= 311.0838. mp = 138-140 °C.
Step 3: 4-(5-Methylthiophen-2-yl)phthalazin-l(2//)-one
2-(Dimethylamino)-3 -hydroxy-3 -(5 -methylthiophen-2-yl)isoindolin- 1 -one (1.1 g, 0.40 mmol), EtOH (4.0 mL), and hydrazine (0.19 mL, 59 mmol) were added into a RBF fitted with a reflux condenser. A nitrogen balloon was attached on top of the condenser. After refluxing overnight, the reaction was cooled to room temperature. An off-white solid precipitated. After cooling to 0 °C, water was added. The solid was filtered off with an aid of water and dried under vacuum to afford a white solid, 4-(5-methylthiophen-2-yl)phthalazin-l(2//)-one (0.82 g, 85% yield).
!H NMR (400 MHz, CDC13) δ 10.57 (s, 1H), 8.50-8.39 (m, 1H), 8.14-8.04 (m, 1H), 7.83- 7.69 (m, 2H), 7.20-7.17 (m, 1H), 6.82-6.71 (m, 1H), 2.47 (s, 3H). 13C NMR (400 MHz,
CDC13) 8 159.9, 142.5, 141.1, 134.3, 133.7, 131.7, 129.4, 128.8, 128.3, 127.1, 126.6,
125.8, 15.4. FT-IR (thin film, cm“1) 2891, 1660, 1334. MS Calcd for Ci3H10N2OS: [M]+
= 242. Found: [M+H]+= 243. HRMS Calcd for Ci3H10N2OS: [M+H]+= 243.0587. Found:
[M+H]+ = 243.0581. mp = 191-194 °C.
Alternatively, starting material 3 was prepared as follows:

The above scheme depicts the process by which intermediate-scale synthesis of thiophene-phthalazinone 5 (shown above) was prepared. Treatment of 50 grams of 3-methylthiophene with z‘-PrMgCl at 66 °C in the presence of catalytic TMP-H resulted in 98% conversion to the reactive species lb with a >40:1 regioisomeric ratio. After cooling to 20 °C, this mixture was added dropwise to a -20 °C slurry of phthalic anhydride in THF to provide keto acid 3 in 94% assay yield. While this intermediate could be crystallized from toluene/heptane, the crude reaction mixture was taken directly in a through -process conversion to the phthalazinone 5. To that end, removal of THF, MTBE, and residual 3-methylthiophene was accomplished through a distillative solvent switch into ethanol. The resulting solution of 3 was exposed to aqueous hydrazine at 80 °C. After 18 hours, the reaction was cooled and the precipitated product was filtered directly at 20 °C. This process provided 82.7 grams of 98.6 wt % thiophene-phthalazinone 5 in a weight-adjusted 85% yield over the two steps.
LCMS Method:
Samples were run on a Agilent model- 1100 LC-MSD system with an Agilent Technologies XDB-C8 (3.5 μ) reverse phase column (4.6 x 75 mm) at 30 °C. The flow rate was constant and ranged from about 0.75 mL/min to about 1.0 mL/min.
The mobile phase used a mixture of solvent A (H2O/0.1% HO Ac) and solvent B
(AcCN/O.1 HOAc) with a 9 min time period for a gradient from 10%> to 90%> solvent B. The gradient was followed by a 0.5 min period to return to 10% solvent B and a 2.5 min 10% solvent B re-equilibration (flush) of the column.
Other methods may also be used to synthesize AMG 900. Many synthetic chemistry transformations, as well as protecting group methodologies, useful in synthesizing AMG 900, are known in the art. Useful organic chemical transformation literature includes, for example, R. Larock, Comprehensive Organic Transformations, VCH Publishers (1989); T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley and Sons (1999); L. Fieser and M. Fieser, Fieser and Fieser’s Reagents for Organic Synthesis, John Wiley and Sons (1994); A. Katritzky and
A. Pozharski, Handbook of Heterocyclic Chemistry, 2nd edition (2001); M. Bodanszky, A. Bodanszky, The Practice of Peptide Synthesis, Springer- Verlag, Berlin Heidelberg (1984); J. Seyden-Penne, Reductions by the Alumino- and Borohydrides in Organic Synthesis, 2nd edition, Wiley- VCH, (1997); and L. Paquette, editor, Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995).
AMG 900 was tested for its ability to reduce or inhibit tumor progression in various cell lines (in-vitro) and multiple solid tumor types (in-vivo), some of which have previously been exposed to and developed resistance to standard-of-care antimitotic agents, including taxanes and vinca alkaloids, as well as to other chemotherapeutic agents. The following Examples and resulting data will illustrate the ability of AMG 900 to treat cancer, including cancer resistant to the presently standard-of-care therapies, including antimitotic agents, such as paclitaxel, and other drugs used in conjunction with chemotherapy, such as doxorubicin. Unless otherwise indicated, the free base form of AMG 900 was used in the Examples described hereinbelow.
The following Examples describe the efforts of identifying and characterizing various crystalline solid state forms of various salts of AMG 900. Some attempts at forming a solid state crystalline form of a given salt failed, as shown in table 1 hereinbelow. To this end, synthesizing and/or forming &isolating a crystalline solid state form of AMG 900 was not, in any way, straightforward or routine. Rather, the ability to prepare and identify a crystalline solid state form of AMG 900 depended upon the particular salt of AMG 900 and/or the crystallization conditions employed.
PAPER
Journal of Medicinal Chemistry (2015), 58(13), 5189-5207
Discovery of N-(4-(3-(2-Aminopyrimidin-4-yl)pyridin-2-yloxy)phenyl)-4-(4-methylthiophen-2-yl)phthalazin-1-amine (AMG 900), A Highly Selective, Orally Bioavailable Inhibitor of Aurora Kinases with Activity against Multidrug-Resistant Cancer Cell Lines
Stephanie Geuns-Meyer*†, Victor J. Cee†, Holly L. Deak†, Bingfan Du†, Brian L. Hodous†, Hanh Nho Nguyen†,Philip R. Olivieri†, Laurie B. Schenkel†, Karina R. Vaida†, Paul Andrews∥, Annette Bak‡, Xuhai Be§, Pedro J. Beltran⊥, Tammy L. Bush⊥, Mary K. Chaves‡, Grace Chung⊥, Yang Dai§, Patrick Eden⊥, Kelly Hanestad⊥, Liyue Huang§, Min-Hwa Jasmine Lin§, Jin Tang∥, Beth Ziegler⊥, Robert Radinsky⊥, Richard Kendall⊥, Vinod F. Patel†, and Marc Payton⊥
†Departments of Medicinal Chemistry, ‡Pharmaceutical Research and Development, §Pharmacokinetics and Drug Metabolism, ∥Molecular Structure, and ⊥Oncology Research, Amgen Inc., 360 Binney Street, Cambridge, Massachusetts 02142, United States, and Amgen Inc., One Amgen Center Drive, Thousand Oaks, California 91320, United States
J. Med. Chem., 2015, 58 (13), pp 5189–5207
DOI: 10.1021/acs.jmedchem.5b00183
ACS Editors’ Choice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes.
Abstract

Efforts to improve upon the physical properties and metabolic stability of Aurora kinase inhibitor14a revealed that potency against multidrug-resistant cell lines was compromised by increased polarity. Despite its high in vitro metabolic intrinsic clearance, 23r (AMG 900) showed acceptable pharmacokinetic properties and robust pharmacodynamic activity. Projecting from in vitro data to in vivo target coverage was not practical due to disjunctions between enzyme and cell data, complex and apparently contradictory indicators of binding kinetics, and unmeasurable free fraction in plasma. In contrast, it was straightforward to relate pharmacokinetics to pharmacodynamics and efficacy by following the time above a threshold concentration. On the basis of its oral route of administration, a selectivity profile that favors Aurora-driven pharmacology and its activity against multidrug-resistant cell lines, 23r was identified as a potential best-in-class Aurora kinase inhibitor. In phase 1 dose expansion studies with G-CSF support, 23r has shown promising single agent activity.
N-(4-(3-(2-Aminopyrimidin-4-yl)pyridin-2-yloxy)phenyl)-4-(4-methylthiophen-2-yl)phthalazin-1-amine (23r)
Applying similar SNAr conditions as for 23b, reaction of 22r and 20a in 2-butanol provided the title compound (2.08 g, 49%) as an off-white solid; mp (DSC) 216 °C.
1H NMR (400 MHz, DMSO-d6) δ ppm 9.36 (s, 1 H) 8.64–8.69 (m, 1 H) 8.41–8.44 (m, 1 H) 8.36–8.40 (m, 1 H) 8.35 (d, J = 5.2 Hz, 1 H) 8.23 (dd, J = 4.8, 2.0 Hz, 1 H) 8.00–8.10 (m, 2 H) 7.91–7.97 (m, 2 H) 7.52 (d, J = 1.0 Hz, 1 H) 7.26–7.33 (m, 3 H) 7.16–7.22 (m, 2 H) 6.74 (br s, 2 H) 2.34 (br s, 3 H).
13C NMR (150 MHz, DMSO-d6) δ 163.81, 160.72, 160.67, 158.68, 151.64, 148.50, 148.36, 147.14, 139.86, 139.24, 137.72, 137.10, 132.61, 131.74, 130.24, 125.27, 124.89, 122.92, 122.83, 122.44, 121.56, 121.52, 119.11, 118.23, 109.93, 15.6
HRMS m/z [M + H]+ Calcd for C28H21N7OS: 504.1601. Found: 504.1607.

| ID, compd name |
AKa |
AK cell assayb |
(nM) |
most potently inhibited non-AKs (nM)c [total kinases in panel] |
admin route |
| 1 (MK-0457/VX-680/tozasertib)(7) |
A/B |
MDA-MB-231 p-HH3(12) |
43 |
FLT3 (6), PLK4 (9), ABL (13), MLCK (15), RET (28); [317](16) |
IV |
| 2 (PHA-739358/danusertib)(8) |
A/B |
MDA-MB-231 p-HH3(12) |
49 |
ABL (25), RET (31), TrkA (31), FGFR1 (47); [35](17) |
IV |
| 3a(AZD1152/barasertib)d,(9) |
B |
MDA-MB-231 p-HH3(12) |
16 |
FLT3 (8), cKIT (17), PDGFRA (38), PDGFRB (41), RET (80); [317](16) |
IV |
| 4 (AT9283)(18) |
A/B |
HCT-116 DNA ploidy |
∼30 |
JAK2 (1), JAK3 (1) Abl (T315I) (4), 9 others ≤10 nM; [230] |
IV |
| 5 (SNS-314)(19) |
A/B |
HCT-116 DNA ploidy(20) |
9 |
TrkB (5), TrkA (12), FLT4 (14), Fms (15), DDR2 (82), Axl (84); [219] |
IV |
| 6 (GSK1070916)(21) |
B |
HCT-116 p-HH3(22) |
20 |
FLT1 (42), TIE2 (59), SIK (70), FLT4 (74), FGFR (78); [328](22) |
IV |
| 7 (ENMD-2076)(23) |
A |
HCT-116 p-AurA |
130 |
FLT3 (2), RET (10), FLT4 (16), SRC (20), TrkA (24), Fms (25); [100] |
PO |
| 8 (CYC116)(24) |
A/B |
A549 p-HH3 |
480 |
VEGFR2 (44), FLT3 (44), CDK2 (390); [23] |
PO |
| 9 (ABT-348)(25) |
A/B |
HCT-116 p-HH3 |
21 |
VEGFR1 (1), FLT3 (1), VEGFR2 (2), CSF-1R (3), PDGFR-α (11); [128] |
PO |
| 10 (AS703569/R763)(26) |
A/B |
A549 p-HH3 |
14 |
cell-based assays: VEGFR2 (11), FLT3 (27), AMPK (201); [10] |
PO |
| 11 (PF-03814735)(27) |
A/B |
MDA-MB-231 p-HH3 |
∼50 |
FLT1 (10), FAK (22), TrkA (30), 17 others ≥90% inh@100 nM; [220] |
PO |
| 12 (MK-5108)(28) |
A |
HeLa S3 ↑p-HH3+ cells |
<1000 |
TrkA (2), ABL (8), FLT4 (12), TrkB (13), VEGFR2 (30); [233] |
PO |
| 13a (MLN8054)(29) |
A |
HCT-116 p-AurA |
34 |
DRAK2 (8), BLK (68), DRAK1 (190), FGR (220); [317](16) |
PO |
| 13b (MLN8237/alisertib)(30) |
A |
HeLa p-AurA |
7 |
%inh@1 μM: EphA2 (111), FGR (97), CAMK2A (95), EphA4 (94); [220] |
PO |
References on AMG 900

| Patent ID |
Date |
Patent Title |
| US2016008316 |
2016-01-14 |
USE OF DIANHYDROGALACTITOL AND ANALOGS OR DERIVATIVES THEREOF IN COMBINATION WITH PLATINUM-CONTAINING ANTINEOPLASTIC AGENTS TO TREAT NON-SMALL-CELL CARCINOMA OF THE LUNG AND BRAIN METASTASES |
| US2016009785 |
2016-01-14 |
NOVEL FUSION MOLECULES AND USES THEREOF |
| US2015266868 |
2015-09-24 |
PHARMACEUTICALLY ACTIVE COMPOUNDS |
| US2015079022 |
2015-03-19 |
USE OF AMG 900 FOR THE TREATMENT OF CANCER |
| US2015072988 |
2015-03-12 |
USE OF N-(4-((3-(2-AMINO-4-PYRIMIDINYL)-2-PYRIDINYL)OXY)PHENYL)-4-(4-METHYL-2-THIENYL)-1-PHTHALAZINAMINE IN COMBINATION WITH HISTONE DEACETYLASE INHIBITORS FOR TREATMENT OF CANCER |
| US8921367 |
2014-12-30 |
Use of AMG 900 for the treatment of cancer |
| US2014163052 |
2014-06-12 |
FUSED TRICYCLIC DUAL INHIBITORS OF CDK 4/6 AND FLT3 |
| US2014127271 |
2014-05-08 |
BLOCK COPOLYMERS FOR STABLE MICELLES |
| US2014113879 |
2014-04-24 |
BLOCK COPOLYMERS FOR STABLE MICELLES |
| US2014114051 |
2014-04-24 |
BLOCK COPOLYMERS FOR STABLE MICELLES |
| Patent ID |
Date |
Patent Title |
| US2014114051 |
2014-04-24 |
BLOCK COPOLYMERS FOR STABLE MICELLES |
| US2014066430 |
2014-03-06 |
AURORA KINASE MODULATORS AND METHOD OF USE |
| US8623885 |
2014-01-07 |
Fused tricyclic dual inhibitors of CDK 4/6 and FLT3 |
| US2012028917 |
2012-02-02 |
Use Of N-(4-((3-(2-Amino-4-Pyrimidinyl)-2-Pyridinyl)Oxy)Phenyl)-4-(4-Methyl-2-Thienyl)-1-Phthalazinamine In The Treatment Of Antimitotic Agent Resistant Cancer |
| US2011263530 |
2011-10-27 |
Aurora Kinase Modulators and Method of Use |
| US8022221 |
2011-09-20 |
Aurora kinase modulators and method of use |
| US7560551 |
2009-07-14 |
Aurora kinase modulators and method of use |
| WO2003055491A1 |
20 Dec 2002 |
10 Jul 2003 |
Astrazeneca Ab |
Substituted quinazoline derivatives as inhibitors of aurora kinases |
| WO2004000833A1 |
19 Jun 2003 |
31 Dec 2003 |
Vertex Pharmaceuticals Incorporated |
Processes for preparing substituted pyrimidines and pyrimidine derivatives as inhibitors of protein kinase |
| WO2004016612A2 |
13 Aug 2003 |
26 Feb 2004 |
Cyclacel Limited |
New purine derivatives |
| WO2004037814A1 |
27 Oct 2003 |
6 May 2004 |
Vertex Pharmaceuticals Incorporated |
Indazolinone compositions useful as kinase inhibitors |
| WO2004039774A2 |
19 May 2003 |
13 May 2004 |
Merck & Co., Inc. |
Mitotic kinesin inhibitors |
| WO2004092607A1 |
30 Mar 2004 |
28 Oct 2004 |
Carbone Lorraine Composants |
Ventilated disc brake pads |
| WO2005113494A2 |
9 May 2005 |
1 Dec 2005 |
Amgen Inc. |
Nitrogenated heterocyclic derivatives as protein kinase modulators and use for the treatment of angiogenesis and cancer |
| EP1702919A1 |
28 Dec 2004 |
20 Sep 2006 |
Banyu Pharmaceutical Co., Ltd. |
Novel 2-heteroaryl-substituted benzimidazole derivative |
| US6919338 |
21 Jun 2001 |
19 Jul 2005 |
Astrazeneca Ab |
Substituted quinazoline derivatives and their use as inhibitors of aurora-2 kinase |
| Citing Patent |
Filing date |
Publication date |
Applicant |
Title |
| WO2008124083A3 * |
3 Apr 2008 |
15 Jan 2009 |
Amgen Inc |
Aurora kinase modulators and method of use |
| WO2009117157A1 * |
19 Mar 2009 |
24 Sep 2009 |
Amgen Inc. |
Aurora kinase modulators and method of use |
| WO2010017240A2 * |
4 Aug 2009 |
11 Feb 2010 |
Amgen Inc. |
Aurora kinase modulators and methods of use |
| WO2010017240A3 * |
4 Aug 2009 |
1 Apr 2010 |
Amgen Inc. |
Aurora kinase modulators and methods of use |
| WO2011031842A1 |
9 Sep 2010 |
17 Mar 2011 |
Amgen Inc. |
N-4 ( – ( ( 3- ( 2 -amino-4 pyrimidinyl) -2 -pyridinyl) oxy) phenyl) -4- (4-methyl-2-thienyl) -1-phthalazinamine for use in the treatment of antimitotic agent resistant cancer |
| WO2012129344A1 |
21 Mar 2012 |
27 Sep 2012 |
Amgen Inc. |
Fused tricyclic dual inhibitors of cdk 4/6 and flt3 |
| WO2015084649A1 |
25 Nov 2014 |
11 Jun 2015 |
Amgen Inc. |
Crystalline forms of n-(4-((3-(2-amino-4-pyrimidinyl) – 2-pyridinyl)oxy)phenyl)-4-(4-methyl-2-thienyl)-1 -phthalazinamine pharmaceutically acceptable salts and uses thereof |
| EP2818170A1 |
9 Sep 2010 |
31 Dec 2014 |
Amgen, Inc |
N-(4-((3-(2-amino-4-pyrimidinyl)-2-pyridin yl)oxy)phenyl)-4-(4-methyl-2-thienyl)-1-phthalazinamine for use in the treatment of antimitotic agent resistant cancer |
| EP2937349A1 |
21 Mar 2012 |
28 Oct 2015 |
Amgen Inc. |
Fused tricyclic dual inhibitors of cdk 4/6 and flt3 |
| US7994185 |
4 May 2009 |
9 Aug 2011 |
Glaxo Smith Kline LLC |
Benzene sulfonamide thiazole and oxazole compounds |
| US8362241 |
|
29 Jan 2013 |
Amgen Inc. |
Inhibitors of PI3 kinase and/or mTOR |
| US8404694 |
19 Mar 2009 |
26 Mar 2013 |
Amgen Inc. |
Aurora kinase modulators and method of use |
| US8415345 |
4 May 2009 |
9 Apr 2013 |
Glaxo SmithKline LLC |
Benzene sulfonamide thiazole and oxazole compounds |
| US8586751 |
10 Jun 2010 |
19 Nov 2013 |
Bristol-Myers Squibb Company |
Nicotinamide compounds useful as kinase modulators |
| US8637500 |
16 Dec 2009 |
28 Jan 2014 |
Amgen Inc. |
Aminopyridine and carboxypyridine compounds as phosphodiesterase 10 inhibitors |
| US8642759 |
31 Jan 2013 |
4 Feb 2014 |
Glaxosmithkline Llc |
Benzene sulfonamide thiazole and oxazole compounds |
| US8772480 |
19 Nov 2012 |
8 Jul 2014 |
Amgen Inc. |
Inhibitors of PI3 kinase and/or mTOR |
| US9126935 |
7 Aug 2009 |
8 Sep 2015 |
Amgen Inc. |
Aurora kinase modulators and methods of use |
| US9233956 |
25 Nov 2013 |
12 Jan 2016 |
Novartis Ag |
Benzene sulfonamide thiazole and oxazole compounds |
///////////945595-80-2, AMG 900, aurora kinase (ARK) inhibitor, treatment of leukemia and solid tumours, AMGEN, 2014, orphan drug designation, U.S. for the treatment of ovarian cancer
CC1=CSC(=C1)C2=NN=C(C3=CC=CC=C32)NC4=CC=C(C=C4)OC5=C(C=CC=N5)C6=NC(=NC=C6)N













Sorry, the comment form is closed at this time.