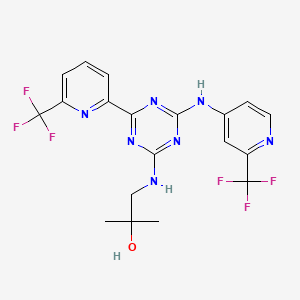
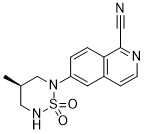
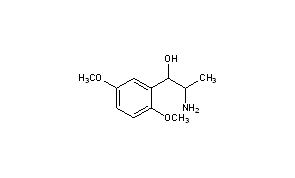
1R,2S-methoxamine, also known as L-erythro-methoxamine
CAS 13699-29-1
- Molecular Weight, 211.26, C11 H17 N O3
HYDROCHLORIDE
(1R,2S)-isomer HCl salt of 1 -(2,5-dimethoxyphenyl)-2-amino-1 -propanol also called as (1R, 2S)methoxamine hydrochloride
CAS 16122-04-6
Used as a pressor agent, as a vasoconstrictor, as a nasal decongestant, in ophthalmology and also found very effective in the treatment of faecal incontinence.
treatment of relief of fecal incontinence and anal itch (pruritis ani) , particularly for patients who have had a major bowel resection and reanastomosis .
Anal or fecal incontinence is the inability to voluntarily control the passage of feces or gas through the anus. It may occur either as fecal soiling or as rare episodes of incontinence for gas or watery stools. It is a very distressing condition that can result in self-inflicted social isolation and despair.
Conventional treatments for fecal incontinence include drug therapy to improve stool consistency, such as morphine, loperamide and codeine phosphate to reduce gut motility, and laxatives to soften stools and relieve constipation. Biofeedback training is another treatment which involves muscle strengthening exercises to improve anal canal resting pressure, and squeeze pressure, and to teach symmetry of anal canal function. The most common form of treatment however, is surgical repair, such as the creation of a neo-sphincter which involves grafting on muscle from other parts of the anus, or a colostomy. (Gastroenterology in Practice, Summer 1995, pl8- 21; Dig Dis 1990; 8:179-188; and The New England Journal of Medicine, April 1992, pl002-1004) . In mild cases of anal leakage, the patient will often try and plug the anus with a ball of cotton wall.
In Gut, 1991, 32, p.345-346 it was reported that two thirds of patients with idiopathic faecal incontinence had a decreased anal resting pressure resulting from an abnormal internal sphincter function. In many incontinent patients, the internal anal sphincter was found to be abnormally thin, while others had an external anal sphincter defect. It has also been reported that in vi tro contractile response of the internal anal sphincter to noradrenaline is decreased in incontinence, (Br. J. Surg. 1992, vol 79, August, p829-832; Digestive Diseases and Sciences, vol 38, no. 11, Nov. 1993, pl961-1969) . A further discussion of the innervation and control of the internal anal sphincter and drugs which can increase or decrease the normal anal resting pressure, is discussed in the text book Coloproctology and the Pelvic Floor (Butterworths) , second edition, 1992, at chapter 3 p37-53; Automic Control of Internal Anal Sphincter; and Journal of Clinical Investigation 1990, 86: p424-429.
In Surgery 1990; 107: p311-315 sodium valproate was found to be useful in the treatment of minor incontinence after ileoanal anastomosis.
It has now surprisingly been found that fecal incontinence and anal itch can be resolved by treatment with α adrenergic agonists, nitric oxide synthase inhibitors, prostaglandins F2α, dopamine, morphine, β-blockers such as propranolol, and 5-Hydroxytryptamine (5-HT) .
This is surprising since it was always thought that once an anal sphincter began functioning abnormally, the patient would require major surgery.
In this way the anal leakage is reduced or eliminated without the patient having to undergo major surgery.
Accordingly in a first aspect of the invention there is provided use of a physiologically active agent selected from an α adrenergic agonist, nitric oxide synthase inhibitor, prostaglandin F2α, dopamine, morphine, β-blockers, and 5- Hydroxytryptamine in the preparation of a medicament for the treatment or prophylaxis of fecal incontinence or anal itch.
The agents of the invention appear to at least partially treat the incontinence by increasing the resting pressure of the internal anal sphincter. Preferred agents are λ adrenergic agonists, nitric oxide synthase inhibitors, and prostaglandins F2α.
Examples of suitable aλ adrenergic agonists are nor- adrenalin, methoxamine, but particularly preferred is phenylephrine .
Examples of suitable F2α prostaglandin are dinoprost and carboprost.
Examples of suitable NO synthase inhibitors are
NG-monnoommeetthhyyll–LL–aarrggiinn:ine (L-NMMA) , and NG-nitro-L-arginine methyl ester ( -NAME)
The medicament can contain a single active agent or a combination of any of the above active agents.
Nitric Oxide (NO) synthase inhibitors such as LNMMA have previously been suggested for the therapeutic treatment of septic shock.
The prostaglandins, along with thromboxanes and leukotrienes are all derived from 20 -carbon polyunsaturated fatty acids and are collectively termed eicosanoids. F2α prostaglandins are derived in vivo from the endoperoxide prostaglandin H2which is in turn derived from leukotrienes. Clinically, F2α prostaglandins such as dinoprost and carboprost are used as uterine stimulants in the termination of pregnancy, missed abortion or the induction of labour.
Phenylephrine (an αx adrenergic agonist) is used as a mydriatic in ophthalmology, and as a decongestant , for example, in cold and flu remedies.
However there has been no suggestion to the inventors knowledge of using any of these active agents to treat fecal incontinence or anal itch. As used herein “fecal incontinence” includes all types of anal leakage from minor leakage or ‘spotting’ through moderate leakage, to major instances of faecal incontinence, and includes neurogenic, active, urge and passive incontinence.
More particularly the class of incontinent patients who will benefit most from the present invention are those with idiopathic incontinence and those whose incontinence is at least partly due to a weakness of either the internal or external anal sphincter, especially those with a normal or low maximum anal pressure and a structurally intact internal anal sphincter muscle, such as with an abnormally thin sphincter. However patients with minor structural damage such as a fragmented sphincter would still benefit from the invention. Not only incontinent patients with a damaged or abnormal internal sphincter can be treated, but also patients with a damaged or abnormal external sphincter since the increase in the internal anal resting tone induced by the invention will compensate for a poorly functioning external sphincter.
Another class of patients who particularly benefit from the invention are post-surgical patients who have had major bowel resection and reanastomosis . For example patients with ileoanal pouch (restorative proctocolectomy) , coloanal (with or without colonic pouch) anostomosis, lower anterior resection, and colectomy with ileorectal anastomosis.
The damage to the sphincter could be caused by trauma, such as experienced in child birth, surgical operations, or road traffic accidents. Furthermore it is also believed that incontinence caused by primary internal anal degeneration can also be relieved by the invention.
Anal leakage also often leads to pruritis of the anus and therefore by reducing or eliminating the leakage, the pruritis or anal itch is also relieved or prevented. Furthermore, as a result of the increased anal resting pressure, the patient no longer has the discomfort of distended anal sphincter muscles.
Methoxamine contains two chiral carbons and thus exists in four isomeric forms. Of all the isomeric forms, the studies revealed (1R,2S)- isomer to be therapeutically active.
US patent 2359707 describes the process for the synthesis of racemic β-(2,5-dimethoxy phenyl)-P-hydroxy-isopropyl amine in neutral, acid salt and its derivative from 2,5- dimethoxy propiophenone by treatment with methylnitrite in diethyl ether medium to obtain 2,5-dimethoxy-a-isonitrosopropiophenone hydrochloride. It is further reduced with palladium on carbon to yield β-(2,5-dimethoxyphenyl)-p-ketoisopropylamine hydrochloride and then with platinum black to get p-(2,5-dimethoxyphenyl)-β- hydroxyisopropyl amine hydrochloride. The described process for di-methoxamine HC1 is not cost-effective, due to the use of two expensive catalysts (platinum black and palladium carbon), solvent diethyl ether and involves more number of steps. The other drawback being it is racemic mixture and cannot be used directly as drug. The process described did not specify the quality of the product.
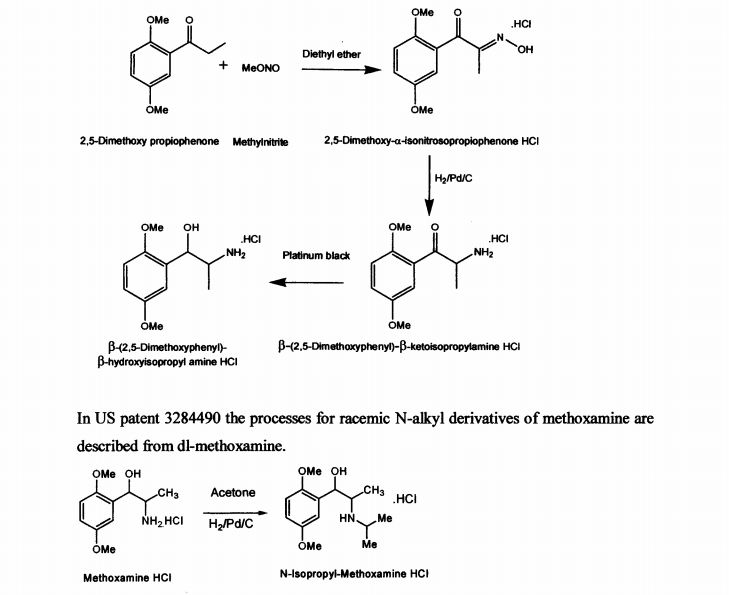
In US patent 3284490 the processes for racemic N-alkyl derivatives of methoxamine are described from dl-methoxamine.
JP 63165348 describes process for production of optically active l-(2,5- dimethoxyphenyl)-2-aminophenol by resolving racemic compound with the use of optically active L-N-acetylleucine as resolving agent. The disadvantages of the process are less yield, low quality and use of expensive naturally occurring amino acid, which prevents from employing this method on commercial scale.
WO 03/055474 A1 discloses mainly, the use of (1R, 2S)-methoxamine in the treatment of faecal incontinence at low doses without local or systemic side effects when used topically. The patent also described the synthesis of (1R, 2S)-methoxamine, from L- alanine, by protecting the amino group using methylchloroformate, converting carboxy
group of the N-protected alanine into an acid chloride insitu followed by reaction with an amine to produce an N-protected (S)-alanine amide and coupling that compound with a brominated 2,5-dimethoxybenzene in the presence of n-butyllithium or a magnesium based reagent to give (S)-amino-l-(2,5-dimethoxy-phenyl)-l-propanone, the amino group of which is protected .The reduction of the N-protected propanone was carried out using dimethylphenylsilane and the protecting group was removed by treatment with potassium hydroxide. Other method adopted in the patent to isolate (1R,2S)methoxamine is by separation of racemic methoxamine using chiral column.
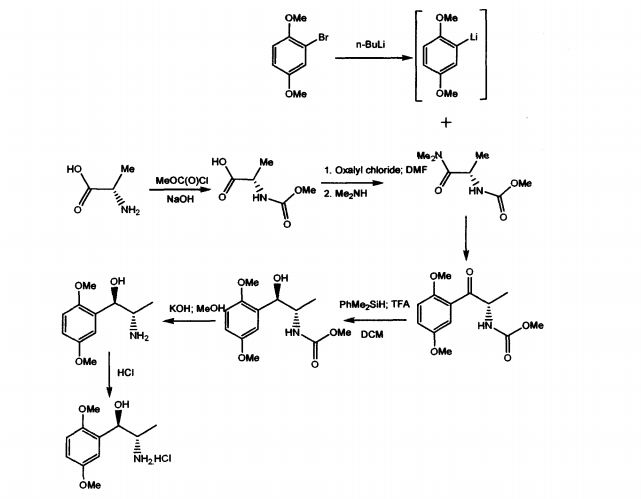
The prior art suffers with some of the disadvantages like using n-butyllithium, which is pyrophoric, expensive and causes hazards to commercial scale. Also, the separation of racemic Methoxamine using chiral column mentioned in the patent can be considered for
isolating small quantities of the required isomer for analytical purposes but cannot be adopted on commercial scale for production of the drug.
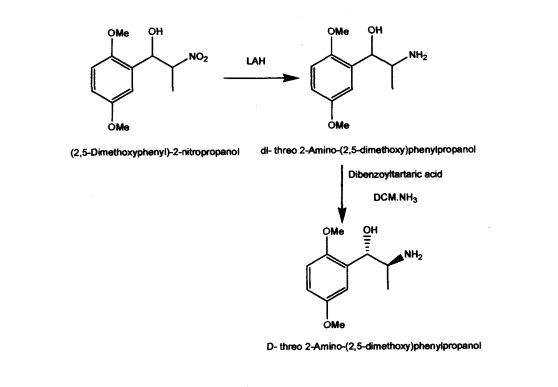
US Patent 5962737 described stereospecific synthesis of the racemic threo isomers of 2- nitro-1 -phenylpropanols by reacting benzaldehyde derivative with nitroalkane in the presence of a tertiary amine and reducing 2-nitro-l-phenylpropanols with lithium aluminium hydride to 2-amino-l-phenylpropanols. Also described is phase transfer resolution of racemic mixtures of 2-amino-l-phenylpropanol and its derivatives into their optically pure isomers by reacting with the mono alkali metal salt of tartaric acid ester in a two phase system of a hydrocarbon and water. The specification further describes optically pure isomer D-threo 2-amino-( 1 -dialkoxy or alkoxy)phenylpropanol by resolution of dl- threo 2-amino-( 1 -dialkoxy or alkoxy)phenylpropanol by using dibenzoyltartaric acid. The synthesis of the product (lS,2S)-threo 2-amino-(l-dialkoxy or alkoxy) phenyl propanol involves the use of expensive and hazardous chemicals like LAH making the process technically and commercially difficult for implementation.
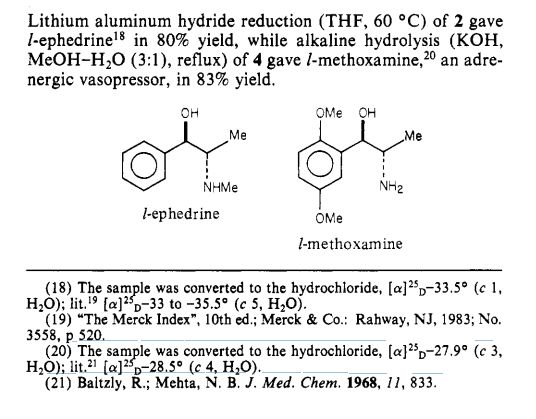
Paper
Journal of the American Chemical Society (1984), 106(16), 4629-30
http://pubs.acs.org/doi/pdf/10.1021/ja00328a062
PATENT
http://www.google.com/patents/EP2275099A1?cl=en
EXAMPLE 3Synthesis of 1R,2S-Methoxamine(S)-N-Methoxycarbonyl alanine
To a stirred solution of L-alanine (300g, 3.37 mol sodium hydroxide (1N, 1800 cm3) at 0°C in an ice bath was added dropwise, over 2 hours, methyl chloroformate (274 cm3, 3.54 mol). The pH of the solution was maintained at 9 by the addition of sodium hydroxide (5N). The reaction mixture was stirred at 0°C for 3 hours whereupon it was acidified to pH 1 by the addition of phosphoric acid solution (15%) and extracted with diethyl ether (5 x 1000 cm3). The combined organic extracts were dried (MgSO4) and concentrated under reduced pressure to yield the product as a viscous green oil (386 g, 78%). 1H NMR (250 MHz; C2HCl3) 1.48 (3H, d, J7.25, CH3), 3.72 (3 H, s, COCH3), 4.40 (1 H, quintet, J7.25, CH), 5.31 (1 H, bs, NH).
(S)-N-Methoxycarbonyl alaninedimethylamide
To a stirred solution of MeOC-alanine (227 g, 1.54 mol) and dimethylformamide (DMF) (25 cm3) in dry dichlorourethane (DCM) (2000 cm3) at 0°C was added dropwise oxalyl chloride (146 cm3, 1.62 mol) over a period of 2 hours. The solution was stirred at 0°C until the evolution of gasses ceased whereupon a basic solution of dimethylamine (676 g, 7.70 mol) in NaOH (3 N, 2000 cm3) was added. The aqueous layer was extracted with diethyl ether (2 x 500 cm3) and the combined organic layers dried (MgSO4) and concentrated under reduced pressure to give the product as a white crystalline solid which required no further purification (230 g, 86%). 1H NMR (250 MHz; C2HCl3) 1.33 (3 H, d, J6.75, CH3), 2.99 3 H, s, OCH3) 3.08, (3 H, s, OCH3), 3.66 (3 H, s, COCH3), 4.66 (H, quintet, J7.00, CH), 5.75 (1 H, d, J5.75, NH).
(S)-2-[(Methoxycarbonyl)amino]-1-(2,5-dimethoxyphenyl)-1-propanone.
To a THF (1000 cm3) solution of bromo-2,5-dimethoxybenzene (55 g, 0.25 mol) at -20°C under nitrogen was addedn-butyl lithium (100 cm3, 2.5 M in hexanes, 0.25 mol). The mixture was stirred at -20°C for 0.75 hours, whereupon a THF (100 cm3) solution of amide (30 g, 0.17 mol) was added via cannula. The solution was stirred at -20°C for 2 hours and was then allowed to warm to room temperature over 1 hour and quenched by the addition of ammonium chloride solution (700 cm3). The solution was diluted with diethyl ether (1000 cm3) and the organic layer was dried (MgSO4) and concentrated under reduced pressure to give a yellow oil. The product was purified by dry flash chromatography on silica (eluant 4:1 hexane/ethyl acetate then 3:2 hexane/ethyl acetate) to give the product as a white crystalline solid (45 g, 98%). 1H NMR (250 MHz; C2HCl3) 1.36 (3 H, d, J7.0, CH3), 3.70 (3 H, s, COCH3), 3.82 (3 H, s, OCH3), 3.92 (3 H, s, OCH3), 5.43 (1 H, quintet, J 7.3, H-2), 5.80 (1 H, bs, NH), 6.94 (1 H, d, J 9.0, ArH), 7.10 (1 H, dd, J 9.0, 3.3, ArH), 7.32 (1 H, d, J 3.3, ArH).
(1R,2S)-2-[(Methoxycarbonyl)amino]-1-(2,5-dimethoxyphenyl)-1-propanol.
To a stirred solution of ketone i.e. (S)-2-[(methoxycarbonyl)amino]-1-(2,5-dimethoxyphenyl)-1-propanone (20 g, 74.9 mmol) and dimethylphenyl silane (10.7 g, 78.6 mmol) in dry DCM (500 cm3) at 0°C in an ice bath was added dropwise trithioroacetic acid (TFA) (50 cm3). The solution was stirred at 0°C for 1 h and then quenched by the addition of sodium hydroxide (500 cm3, 1 N). The organic layer was dried and concentrated under reduced pressure to give a yellow oil which solidified on standing. This solid was crystallized from ether/hexane to give the product as a white crystalline solid (15.6 g, 75%).1H NMR (250 MHz; C2HCl3) 1.03 (3 H, d, J7.0, CH3), 3.04 (1 H, d, J4.3, OH), 3.68 (3 H, s, COCH3), 3.78 (3 H, s, OCH3), 3.80 (3 H, s, OCH3), 3.94-3.99 (1 H, m, H-2), 5.05-5.15 (2 H, m, H-1 and NH), 6.72-6.85 (2 H, m, ArH) 6.97 (1 H, d, J 2.0, ArH).
(1,R,2S)-Methoxamine.
To a stirred solution of methoxycarbonyl (MeOC) protected alcohol i.e. (1R,2S)-2-[(methoxycarbonyl)amino]-1-(2,5-dimethoxyphenyl)-1-propanol (4.0 g, 14.9 mmol) in methanol (175 cm3) was added a solution of KOH (4.06 g, 72.8 mmol in water (60 cm3). The solution was cooled and acidified with phosphoric acid (15% v/v). The solution was extracted with DCM (2 x 50 cm3) and the aqueous layer basified by the addition of K2CO3. The aqueous layer was extracted with diethyl ether (5 x 50 cm3) and the combined ethereal extracts dried (MgSO4) and concentrated under reduced pressure to give the product as a clear yellow oil (1.9 g, 61%), 1H NMR (250 MHz; C2HCl3) 0.84 (3 H, d, J 7.0, CH3), 3.19-3.22 (1 H, m, H-2), 3.71 (6 H, s, 2 x OCH3), 4.67 (1 H, d, J 5.0, H-1), 6.66-6.72 (2 H, m, ArH), 6.92 (1 H, d, J 2.5, ArH).
(1R, 2S)-Methoxamine hydrochloride.
To an ice cooled solution of (1R,2S)-methoxamine (1.9 g, 9.00 mmol) in anhydrous diethyl ether (30 cm3) was passed a stream of dry HCl gas for 45 mins. The resultant precipitate was filtered by suction, washed with cold diethyl ether and dried under nitrogen to yield the title compound as a white solid. (1.5 g, 68%). 1H NMR (250 MHz; [C2H3]2SO) 0.89 (3 H, d, J 6.8, CH3), 3.37-3.42 (1 H,m,H-2), 3.71 (3 H, s, OCH3), 3.75 (3 H, s, OCH3), 5.12 (1 H, s, H-1), 5.92 (1 H, d, J 4.3, OH), 6.84 (1 H, dd, J 8.8, 3.0, ArH), 6.92-7.00 (2 H, m, ArH); HPLC.
Analytical Method for the Analysis of Methoxamine
The following method was used to analyse methoxamine samples.
Method
-
Column : Cyclobond I RSP 250 x 4.6 mm Column temperature : 23°C Mobile phase : 0.1% Tetraethylammonium pH 4.1* 95%v/v : Acetonitrile 5%v/v Flow rate : 0.6 ml/min Solution
Concentration :5 mg/l Injection volume : 2.5 µl to 20 µl Detection : UV 230 nm *Tetraethylammonium acetate pH 4.1 was prepared fresh daily.
Example 2 above allows the complete assignment of the methoxamine isomers as shown below:


PATENT
INDIAN 1020/CHE/2011
BY


The Managing Director of Malladi Drugs & Pharmaceuticals, Prashant Malladi (left), with the Chief Executive Officer, V. N. Gopalakrishnan

V.N Gopalakrishnan
CEO at Malladi Drugs & Pharmaceuticals Ltd

Prabhakaran Ranganathan
Vice President (Operations) at Malladi Drugs and Pharmaceuticals Limited

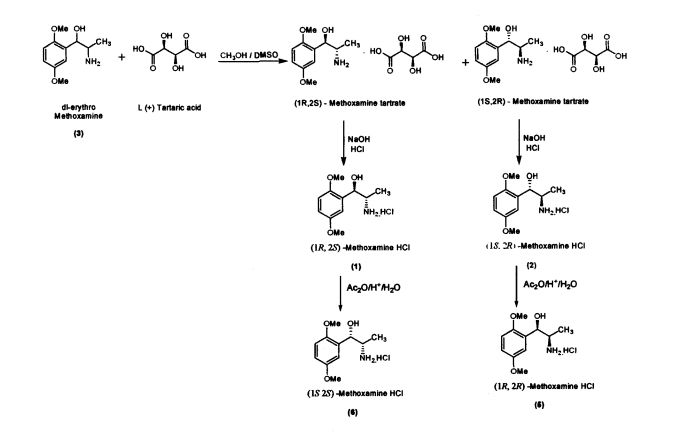
The present invention further provides an improved process for the preparation of (JS, 2S)-Methoxamine HC1 of formula (6) from (1R, 2S)-methoxamine by treating with acetic anhydride in toluene medium followed by acid hydrolysis and basification to obtain (IS, 2S)-Methoxamine base which is further acidified to form (1S,2S)- Methoxamine HC1 (6).
The present invention further provides an improved process for the preparation of (1R, 2R)-Methoxamine HC1 of formula (5) from its diastereomer (1S, 2R)-methoxamine HC1 of formula (2) by treating with acetic anhydride in toluene medium followed by acid hydrolysis and basification to obtain (1R, 2R)-Methoxamine base which is further acidified to form (1R, 2R)-Methoxamine HC1 (5).
The following examples illustrate the invention.
EXAMPLES
Example 1
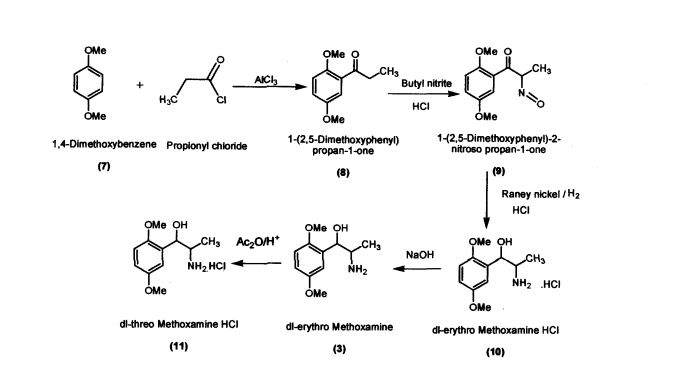
Preparation of l-(2,5-Dimethoxyphenyl)propan-l-one (8)
Aluminium chloride (127.4 g; 0.955 mol) was added to dichloromethane (420 mL) in a round bottomed flask under nitrogen atmosphere. The reaction mixture was cooled to -5 °C; 1,4-dimethoxybenzene (100 g; 0.724 mol) was added slowly within 15-30 minutes. Propionic chloride (87 g; 0.94 mol) dissolved in dichloromethane (245 mL) was added slowly within 2 hours. The reaction mass was allowed to stir for 2 hours and then was quenched in crushed ice (1 kilo) and HC1 (75 mL) at 0 – 5 °C. Separated the layers and the organic layer was washed with 5% sodium hydroxide solution, dried and concentrated (140 g; colorless liquid); Purity by HPLC : 99.04%
Spectroscopic interpretation
The structure of the product, l-(2,5-Dimethoxyphenyl)propan-l-one was confirmed with the help of the following spectroscopic data.
a) IR (cm-1) (KBr)
Aromatic C-H stretch at 3071, aliphatic C – H stretch at 2938, C = O stretch at 1674, benzenoid bands at 1609 and 1584, C – O stretch at 1223, C – H out of plane bending of tri-substituted benzene ring at 814,719.
b) 1H NMR(CDCb, 300 MHz) (δH)
1.16 (3H, t, -CH2-CH3), 3.0 (2H, q, -CH2-CH3), 3.78 (3H, s, -OCH3), 3.85 (3H, s, -OCH3), 6.83 – 7.72 (3H, m, aromatic protons)
c) 13C NMR (CDCb, 300 MHz) (δC)
8.44 (-CH2-CH3), 37.03 (-CH2-CH3), 55.74 (-OCH3), 56.01 (-OCH3), 113.09 – 153.41 (aromatic carbons), 202.96 (C=O)
d) Mass spectrum (ESI, methanol)
[M+Na]+ at m/z 217 (9), [M+H]+ at m/z 195 (100).
Example 2
Preparation of l-(2,5-Dimethoxyphenyl)-2-nitrosopropan-l-one (9) l-(2,5-Dimethoxyphenyl)propan-l-one (100 g; 0.515 mol) was added to dichloromethane (660 mL) in a round bottomed flask under nitrogen atmosphere. Butylnitrite (46.6 g; 0.52 mol) was slowly added in about 30 minutes at 30 – 35 °C. Diethyl ether (60.2 mL) was added to the reaction mixture and dry HC1 gas was purged for about 4 hours at 30 – 35 °C. The reaction mass was maintained for 12 hours and then concentrated under vacuum The residue obtained (60 g; Pale yellow crystalline powder); Purity by HPLC: 99.81%; mp: 104-107 °C
Spectroscopic interpretation
The structure of the product, l-(2,5-Dimethoxyphenyl)-2-nitrosopropan-l-one was confirmed with the help of the following spectroscopic data
a) IR (cm1) (KBr)
O-H stretch at 3250 (broad), aromatic C-H stretch at 3024, aliphatic C – H stretch at 2934, C = O stretch at 1688, C = N stretch at 1645, benzenoid bands at 1589 and 1504, C-O stretch at 1231, C-H out of plane bending of tri-substituted benzene ring at 745,702.
b) 1H NMR(CDCb, 300 MHz) (δh)
2.07 (3H, s, -C-CH3), 3.72 (3H, s, -OCH3), 3.76 (3H, s, -OCH3), 6.84-6.99 (3H, m, aromatic protons), 8.89 (1H, bs, OH)
c) 13C NMR (CDCb, 300 MHz) (δC)
9.16 (-C-CH3), 55.81 (-OCH3), 56.34 (-OCH3), 113.09 – 153.27 (aromatic carbons), 157.07 (C=N-OH); 193.32 (CO)
d) Mass spectrum (ESI, methanol) [M+H]+ at m/z 224 (100)
Example 3
Preparation of dl-erythro-methoxamine HC1 (10)
Raney nickel (50 g); iso-propyl alcohol (250 mL) were added to the autoclave. l-(2,5- Dimethoxyphenyl)-2-nitrosopropan-1 -one (100 g; 0.448 mol) was added slowly at 50 – 55 °C by simultaneously purging the flask with hydrogen at 2-3 Kilo pressure. When hydrogen consumption ceases, the catalyst was filtered and the filtrate was concentrated. iso-Propyl alcohol (200 mL) was added to the concentrated mass followed by acidification with HC1 to obtaindl-erythro-methoxamine HC1 (70 g; white crystalline solid)
Spectroscopic interpretation
The structure of the product, dl-erythro-methoxaxmne HC1 was confirmed with the help of the following spectroscopic data.
a) IR (cm1) (KBr)
O-H stretch at 3409, aromatic C-H stretch at 3010, aliphatic C – H stretch at 2914, HN-H str. at 2574 and 2467, benzenoid bands at 1615 and 1569, C-N stretch at 1279, C-O stretch at 1216, C-H out of plane bending of 1,2,4-tri- substituted benzene ring at 812.
b) 1H NMR (DMSO-d6, 300 MHz) (δH)
1.0 (3H,d, -CH-CH3), 3.74 (3H, s, -OCH3), 3.77 (3H, s, -OCH3), 4.89 (1H, q, -CH-CH3),6.1 (1H, d, -CH-OH), 6.87-7.01 (3H, m, aromatic protons), 8.06 (3H, bs, HN-H) The -OH proton appears to have exchanged with the solvent.
c) 13C NMR (DMSO-d6, 300 MHz) (δc)
14.75 (-CH-CH3), 52.12 (-OCH3), 55.70 (-OCH3), 55.70 (-CH-CH3), 67.25 (CH-OH), 111.89 – 153.16 (aromatic carbons)
d) Mass spectrum (ESI, methanol)
[M+H)+ at m/z 212 (100), [M-H2O]+ at m/z 194 (56).
Example 4
Preparation of(JR,2S)-Metboxamine HC1 (1) and (1S, 2R)-Methoxamine HC1 (2) dl-erythro-methoxamine HC1 (117g; 0.47 mol) was dissolved in water (350 mL) at 30-35 °C. The clear solution obtained was basified using 50% sodium hydroxide solution. dl-erythro-Methoxaumne (3) was extracted into dichloromethane (150 mL) and concentrated. Mixture of methanol/DMSO (4:1; 1650 mL) was added and the mass was heated to 50 °C. L-(+)-Tartaric acid (71.1g; 0.47mol) was added slowly and the temperature of the mass was further raised to 70 °C for complete dissolution. The mass was cooled to 35 °C and maintained for 48 hours. (IR,2.S)-Methoxamine tartrate complex (80 g) precipitated was filtered. From the filtrate on concentration was obtained (1S,2R)- methoxamine tartrate complex (82 g) (IR,25)-Methoxamine tartrate complex was added to water (250 mL) at 35 °C, basified to 12 – 13 pH with 50% sodium hydroxide solution. Dichloromethane (200 mL) was added and stirred for 30 min. Separated the org layer, dried over sodium sulphate and concentrated completely under vacuum at 45° C. Iso-Propyl alcohol (150 mL) was added, charcaolized and filtered. The clear filtrate was acidified with 20%IPA HC1 to yield (1R, 2S)-Methoxamine HC1 which was filtered and dried (48 g); White crystalline powder; Purity by HPLC : 100%; Chiral purity : 100 %; mp : 172-175 °C; [α]D: -47.94° (c = 2% in MeOH)
Spectroscopic interpretation
The structure of the product, (1R,2S)-Methoxamine HC1 was confirmed with the help of the following spectroscopic data.
a) IR (cm1) (KBr)
O-H stretch at 3300, aromatic C-H stretch at 3065, aliphatic C-H stretch at 2938, HN-H str. at 2693 and 2580, benzenoid bands at 1609 and 1578, C-N stretch at 1277, C-O stretch at 1217, C-H out of plane bending of 1,2,4-tri- substituted benzene ring at 818.
b) 1H NMR (DMSO-d6 300 MHz) (δH)
0.91 (3H,d, -CH-CH3), 3.71 (3H, s, -OCH3), 3.75 (3H, s, -OCH3), 5.14 (1H, m, -CH- NH3+), 5.95 (1H, d, -CH-OH), 6.83-7.01 (3H, m, aromatic protons), 8.25 (3H, bs, HN-H) The -OH proton appears to have exchanged with the solvent.
c) 13C NMR (DMSO-d6, 300 MHz) (δC)
II. 44 (-CH-CH3), 49.22 (-OCH3), 55.24 (-OCH3), 55.70 (-CH-CH3), 66.49 (CH-OH),
III. 41 – 153.03 (aromatic carbons)
d) Mass spectrum (ESI, methanol)
[M+H]+ at m/z 212 (100), [M-H2O]+ at m/z 194 (15).
(IS, 2i?)-Methoxamine tartrate complex was added to water (275 mL) at 35 °C, basified
to 12 – 13 pH with 50% sodium hydroxide solution. Dichloromethane (250 mL) was added and stirred for 30 min. Separated the organic layer, dried over sodium sulphate and concentrated completely under vacuum at 45 °C. Iso-Propyl alcohol (175 mL) was added, charcaolized and filtered. The clear filtrate was acidified with 20%IPA HC1 to yield (1S, 2R)-Methoxamine HC1 which was filtered and dried (51 g) White crystalline powder; Purity by HPLC : 99.99%; Chiral purity . 100 %; mp . 172-175 °C;[α]D : + 47.9° (c = 2% in MeOH)
Spectroscopic interpretation
The structure of the product, (1S, 2R)-Methoxamine HC1 was confirmed with the help of the following spectroscopic data.
a) m (cm1) (KBr)
O-H stretch at 3265, aromatic C-H stretch at 3059, aliphatic C-H stretch at 2997, HN-H str. at 2658 and 2567, benzenoid bands at 1611 and 1587,
C-N stretch at 1294, C-O stretch at 1217, C-H out of plane bending of 1,2,4-tri- substituted benzene ring at 818.
b) 1H NMR (DMSO-d6,300 MHz) (δH)
0.91 (3H,d, -CH-CH3), 3.71 (3H, s, -OCH3), 3.75 (3H, s, -OCH3), 5.14 (1H, m, -CH- NH3+), 5.97 (1H, d, -CH-OH), 6.83-7.01 (3H, m, aromatic protons), 8.19 (3H, bs, HN-H) The -OH proton appears to have exchanged with the solvent.
c) 13C NMR (DMSO-d6,300 MHz) (δc)
II. 46 (-CH-CH3), 49.18 (-OCH3), 55.23 (-OCH3), 55.68 (-CH-CH3), 66.45 (CH-OH),
III. 42 – 153.02 (aromatic carbons)
d) Mass spectrum (ESI, methanol)
[M+H]+ at m/z 212 (100), [M-H2O]+ at m/z 194 (15).
Example 5
Preparation of dl-threo-methoxamine HC1 (11)
dl-erythro-methoxamine HC1 (120g; 0.48 mol) was dissolved in DM water (500 mL) at 30 – 35 °C and cooled to 10 – 15 °C. The clear solution was basified using 50 % sodium hydroxide solution and extracted in dichloromethane (250 mL). The organic layer was separated and concentrated under vacuum. The residue thus obtained was dissolved in toluene (200 mL) and was added slowly to acetic anhydride (120 g; 1.17mol) at 65 – 70 °C. The reaction mass was maintained under stirring and further cooled to 10 – 20 °C. Conc.Sulphuric acid (57.6g; 0.58mol) was added to the reaction mass slowly by maintaining the reaction mass at 10 – 200 C. The reaction mass was heated to 35 – 400 C for 3 hours and concentrated under vacuum at below 80 °C.
The reaction mass was cooled to 10 – 15 °C and was dissolved in DM water (250 mL). The mass was maintained for 3 h at reflux temperature and again cooled to 10 – 15 °C.
The pH was adjusted to 12 – 13 using 50% sodium hydroxide solution and extracted the d/-threo-Methoxamine base in dichloromethane (250 mL). Separated the organic layer and concentrated under vacuum. The concentrated mass was triturated with iso-Propyl alcohol (150 mL); acidified using 20% HC1 in iso-propyl alcohol. Distilled the iso- propyl alcohol completely to the final traces and acetone (300 mL) was added. The material precipitated, crude dl-threo-methoxamine HC1 was filtered. (85 g) Off white powder; Purity by HPLC: 99.4%; mp: 221-223 °C Spectroscopic interpretation
The structure of the product, di-threo-methoxamine HC1 was confirmed with the help of the following spectroscopic data.
a) IR (cm”1) (KBr)
O-H stretch at 3401, aromatic C-H stretch at 3005, aliphatic C-H stretch at 2924, HN-H str. at 2581 and 2490, benzenoid bands at 1609 and 1578, C-N stretch at 1277, C-0 stretch at 1215, C-H out of plane bending of 1,2,4-tri- substituted benzene ring at 802.
b) NMR (DMSO-d6,300 MHz) (δH)
1.2 (3H,d, -CH-CHs), 3.72 (3H, s, -OCH3), 3.75 (3H, s, -OCH3), 4.87 (1H, q, -CH-CH3),6.3 (1H, d, -CH-OH), 6.83-6.99 (3H, m, aromatic protons), 8.03 (3H, bs, HN-H) The -OH proton appears to have exchanged with the solvent.
c) 13C NMR (DMSO-d6, 300 MHz) (δC)
14.76 (-CH-CH3), 52.15 (-OCH3), 55.89 (-OCH3), 67.34 (CH-OH), 111.96 – 153.21 (aromatic carbons)
d) Mass spectrum (ESI, methanol)
[M+H]+ at m/z 212 (100), [M-H2O]+ at m/z 194 (52).
Example 6
Preparation of (1S,2S)- Methoxamine HC1 (6)
(IR, 2S)-Methoxamine HC1 (120 g; 0.48 mol) was dissolved in DM water (500 mL) at 30 -35 °C and cooled to 10 – 15 °C. The clear solution was basified using 50 % sodium hydroxide solution and extracted in dichloromethane (250 mL). The organic layer was separated and concentrated under vacuum. The residue thus obtained was dissolved in toluene (200 mL) and was added slowly to acetic anhydride (120 g; 1.17 mol) at 65 – 70 °C. The reaction mass was maintained under stirring and further cooled to 10 – 20 °C. Conc.sulphuric acid (57.6 g; 0.58 mol) was added to the reaction mass slowly by maintaining the reaction mass at 10 – 20 °C. The reaction mass was heated to 35 – 40 °C for 3 hours and concentrated under vacuum at below 80 °C.
The reaction mass was cooled to 10-15°C and was dissolved in DM water (250 mL). The mass was maintained for 3 h at reflux temperature and again cooled to 10 – 15 °C. The pH was adjusted to 12-13 using 50% sodium hydroxide solution and extracted the (1S, 2S)-Methoxamine base in dichloromethane (250 mL). Separated the organic layer and concentrated under vacuum The concentrated mass was triturated with iso-Propyl alcohol (150 mL); acidified using 20% HC1 in iso-propyl alcohol. Distilled the iso- propyl alcohol completely to the final traces and acetone (300 mL) was added. The material precipitated, crude (IS, 2S)-methoxamine HC1 was filtered. (86 g); White crystalline powder; Purity by HPLC . 99.8%; Chiral purity : 99.7%; mp : 172-175 °C; [α]D: + 30.739° (c = 2% in MeOH)
Spectroscopic interpretation
The structure of the product, (IS, 2S)-methoxamine HC1 was confirmed with the help of the following spectroscopic data.
a) IR (cm1) (KBr)
O-H stretch at 3356, aromatic C-H stretch at 3080, aliphatic C-H stretch at 2999, HN-H str. at 2641 and 2583, benzenoid bands at 1611 and 1506, C-N stretch at 1302, C-O stretch at 1229, C-H out of plane bending of 1,2,4-tri- substituted benzene ring at 812.
b) 1H NMR (DMSO-d6 300 MHz) (δH)
1.04 (3H,d, -CH-CH3), 3.72 (3H, s, -OCH3), 3.75 (3H, s, -OCH3), 4.90 (1H, m, -CH- CH3),6.07 (1H, d, -CH-OH), 6.84-7.01 (3H, d, aromatic protons), 8.15 (3H, bs, HN-H)
The -OH proton appears to have exchanged with the solvent.
c) 13C NMR (DMSO-d6, 300 MHz) (δC)
14.75 (-CH-CH3), 52.18 (-OCH3), 55.21 (-OCH3), 55.69 (-CH-CH3), 67.32 (CH-OH), 111.38 -153.01 (aromatic carbons)
d) Mass spectrum (ESI, methanol)
[M+H]+ at m/z 212 (100), [M-H2O]+ at m/z 194 (48).
Example 7
Preparation of (1R, 2R)-Methoxamine HC1 (5)
(IS, 2R)Methoxamine HC1 (120g; 0.48 mol) was dissolved in DM water (500 mL) at 30 – 35 °C and cooled to 10 – 15 °C. The clear solution was basified using 50 % sodium hydroxide solution and extracted in dichloromethane (250 mL). The organic layer was separated and concentrated under vacuum. The residue thus obtained was dissolved in toluene (200 mL) and was added slowly to acetic anhydride (120 g; 1.17mol) at 65 – 70 °C. The reaction mass was maintained under stirring and further cooled to 10 – 20 °C. Cone.Sulphuric acid (57.6g; 0.58mol) was added to the reaction mass slowly by maintaining the reaction mass at 10 – 20 °C. The reaction mass was heated to 35 – 40 °C for 3 hours and concentrated under vacuum at below 80 °C.
The reaction mass was cooled tol0-15°C and was dissolved in DM water (250 mL). The mass was maintained for 3 h at reflux temperature and again cooled to 10 – 15 °C. The pH was adjusted to 12-13 using 50% sodium hydroxide solution and extracted the (IR, 2i?)-Methoxamine base in dichloromethane (250 mL). Separated the organic layer and concentrated under vacuum. The concentrated mass was triturated with iso-Propyl alcohol (150 mL); acidified using 20% HC1 in iso-propyl alcohol Distilled the iso- propyl alcohol completely to the final traces and acetone (300 mL) was added. The material precipitated, crude (1R, 2R)-methoxamine HC1 was filtered. (90 g) White crystalline powder; Purity by HPLC: 99.1%, Chiral purity. 100%; mp: 172-175 °C;[α]D: -29.04° (c – 2% in MeOH)
Spectroscopic interpretation
The structure of the product, (1R, 2R)methoxamine HC1 was confirmed with the help of the following spectroscopic data.
a) IR (cm1) (KBr)
O-H stretch at 3356, aromatic C-H stretch at 3078, aliphatic C-H stretch at 2999, HN-H str. at 2619 and 2500, benzenoid bands at 1611 and 1508, C-N stretch at 1302, C-O stretch at 1229, C-H out of plane bending of 1,2,4-tri- substituted benzene ring at 812.
b) 1H NMR(DMSO-d6 300 MHz) (δH)
I. 04 (3H,d, -CH-CHa), 3.72 (3H, s, -OCH3), 3.75 (3H, s, -OCH3), 4.90 (1H, m, -CH- CH3),6.07 (1H, d, -CH-OH), 6.83-7.01 (3H, d, aromatic protons), 8.13 (3H, bs, HN-H) The -OH proton appears to have exchanged with the solvent.
c) 13C NMR (DMSO-d6 300 MHz) (δe)
II. 41 (-CH-CH3), 52.16 (-OCH3), 55.22 (-OCH3), 55.70 (-CH-CH3), 67.32 (CH-OH), III. 39-153.15 (aromatic carbons)
d) Mass spectrum (ESI, methanol)
[M+H]+ at m/z 212 (100), [M-H2O]+ at m/z 194 (44).
PATENT
http://www.google.com/patents/US8491931
(1,R,2S)-Methoxamine
To a stirred solution of methoxycarbonyl (MeOC) protected alcohol i.e. (1R,2S)-2-[(methoxycarbonyl)amino]-1-(2,5-dimethoxyphenyl)-1-propanol (4.0 g, 14.9 mmol) in methanol (175 cm3) was added a solution of KOH (4.06 g, 72.8 mmol in water (60 cm3). The solution was cooled and acidified with phosphoric acid (15% v/v). The solution was extracted with DCM (2×50 cm3) and the aqueous layer basified by the addition of K2CO3. The aqueous layer was extracted with diethyl ether (5×50 cm3) and the combined ethereal extracts dried (MgSO4) and concentrated under reduced pressure to give the product as a clear yellow oil (1.9 g, 61%), 1H NMR (250 MHz; C2HCl3) 0.84 (3H, d, J 7.0, CH3), 3.19-3.22 (1H, m, H-2), 3.71 (6H, s, 2×OCH3), 4.67 (1H, d, J 5.0, H-1), 6.66-6.72 (2H, m, ArH), 6.92 (1H, d, J 2.5, ArH).
(1R,2S)-Methoxamine hydrochloride
To an ice cooled solution of (1R,2S)-methoxamine (1.9 g, 9.00 mmol) in anhydrous diethyl ether (30 cm3) was passed a stream of dry HCl gas for 45 mins. The resultant precipitate was filtered by suction, washed with cold diethyl ether and dried under nitrogen to yield the title compound as a white solid. (1.5 g, 68%). 1H NMR (250 MHz; [C2H3]2SO) 0.89 (3H, d, J 6.8, CH3), 3.37-3.42 (1H,M,H-2), 3.71 (3H, s, OCH3), 3.75 (3H, s, OCH3), 5.12 (1H, s, H-1), 5.92 (1H, d, J 4.3, OH), 6.84 (1H, dd, J 8.8, 3.0, ArH), 6.92-7.00 (2H, m, ArH); HPLC.



//1R,2S-methoxamine
RACEMIC
 |
|
Title: Methoxamine
CAS Registry Number: 390-28-3
CAS Name: a-(1-Aminoethyl)-2,5-dimethoxybenzenemethanol
Additional Names: a-(1-aminoethyl)-2,5-dimethoxybenzyl alcohol; 2-amino-1-(2,5-dimethoxyphenyl)-1-propanol; b-hydroxy-b-(2,5-dimethoxyphenyl)isopropylamine; b-(2,5-dimethoxyphenyl)-b-hydroxyisopropylamine; 2,5-dimethoxynorephedrine
Molecular Formula: C11H17NO3
Molecular Weight: 211.26
Percent Composition: C 62.54%, H 8.11%, N 6.63%, O 22.72%
Literature References: a1-Adrenergic agonist. Prepn: Baltzly et al., US 2359707 (1944 to Burroughs Wellcome). Metabolism: A. Klutch, M. Bordun, J. Med. Chem. 10, 860 (1967). Clinical pharmacology: N. T. Smith, C. Whitcher, Anesthesiology 28, 735 (1967); P. D. Snashall et al., Clin. Sci. Mol. Med. 54, 283 (1978). HPLC determn in plasma: I. A. Al-Meshal et al., J. Liq. Chromatogr. 12, 1589 (1989). Therapeutic use: P. M. C. Wright et al., Anesth. Analg. 75, 56 (1992); L. Cabanes et al., N. Engl. J. Med. 326, 1661 (1992). Comprehensive description: A. M. Al-Obaid, M. M. El-Domiaty, Anal. Profiles Drug Subs. 20, 399-431 (1991).
Derivative Type: Hydrochloride
CAS Registry Number: 61-16-5
Trademarks: Vasoxine (Burroughs Wellcome); Vasoxyl (Burroughs Wellcome); Vasylox (Burroughs Wellcome)
Molecular Formula: C11H17NO3.HCl
Molecular Weight: 247.72
Percent Composition: C 53.33%, H 7.32%, N 5.65%, O 19.38%, Cl 14.31%
Properties: Crystals, mp 212-216°. pKa (25°C) 9.2. Very sol in water: One gram dissolves in 2.5 ml water, in 12 ml ethanol. Practically insol in ether, benzene, chloroform. pH of a 2% aq soln between 4.5 and 5.5.
Melting point: mp 212-216°
pKa: pKa (25°C) 9.2
Therap-Cat: Antihypotensive.
Keywords: a-Adrenergic Agonist; Antihypotensive.
|