











Areas of discussion included how expectations for raw material control are evolving within changing regulatory and business paradigms including quality by design (QbD), counterfeiting, complex supply chains, and sourcing changes. discussed risk assessment and mitigation strategies along with supplier risk management plans.

Regulatory Considerations
the lack of a consistent definition of raw materials in regulations pertaining to the pharmaceutical industry. In its Q7 guideline, the International Conference on Harmonisation of Technical Requirements for the Registration of Pharmaceuticals for Human Use (ICH) defines raw materials as “starting materials, reagents, and solvents intended for use in the production of intermediates or APIs.” However, the term as defined by different speakers could cover a wide range of materials including the following:
• starting or source materials (cell lines, viral or bacterial stocks, media components, chemicals, tissues, serum, water)
• in-process materials (resins, buffers, filters, column housings, tubing, reagents)
• excipients
• packaging components, both primary and secondary (syringes, vials, stoppers, plungers, crimps, boxes, trays, and labels)
• device/delivery components (pen/ injector components, IV bags, filters). Some regulations directly consider the control of raw materials, but they are not comprehensive and are scattered among the US Code of Federal Regulations (CFR), ICH, and other regulations/guidances. Although the regulations are not extensive, the need to control raw materials was clear from all presenters and is implicit in the sources cited below:
• 21 CFR 610.15 regarding constituents
• 21 CFR 211.80 regarding components and containers/closures
• 21 CFR 211.110 regarding control of in-process materials • ICH Q5A/D for cell substrates and viral safety
• ICH Q7 discussing the need to control materials with appropriate specifications
• ICH Q10 stating that a biomanufacturer is responsible for the quality of purchased materials
• the US bill “Country-of-Origin Labeling for Pharmaceutical Ingredients,” proposed in September 2008
• QbD principles requiring an understanding of the criticality of quality attributes for raw materials and their effect on processes and products.
Developing Control
Strategies Control of raw materials is essential to maintaining safety. Thorough knowledge of raw materials can mitigate the potential for contamination derived from such sources as microbes, chemicals, prions, and pyrogens. Raw material control for safety also includes identification — being able to verify that you have received the correct material — because the presence of an incorrectly identified material in a manufacturing process could compromise safety.

Control of raw materials is essential to ensure lot-to-lot consistency because variation in them can directly affect the variation of both product and process. So manufacturers must understand the critical material attributes (CMAs) of their raw materials and which of those affect variability — as well as how to control that variability.
You must show that you are using appropriate analytical methods to characterize raw materials. Raw materials such as polyethylene glycol (PEG) isomers, trace materials in media and water, container and closure materials, and chromatography resins all have the potential to affect lot-to-lot consistency. An effective raw material control program will also ensure consistent supplies.
A single source for a vital raw material can be a significant financial and quality-assurance risk. If a supplier goes out of business or experiences quality problems, can that raw material be obtained elsewhere? Has a second source been qualified in case the primary source is no longer available? Does the second source have the capacity to meet your needs? A QbD approach to raw material control requires that you understand the impact on your product’s critical quality attributes.
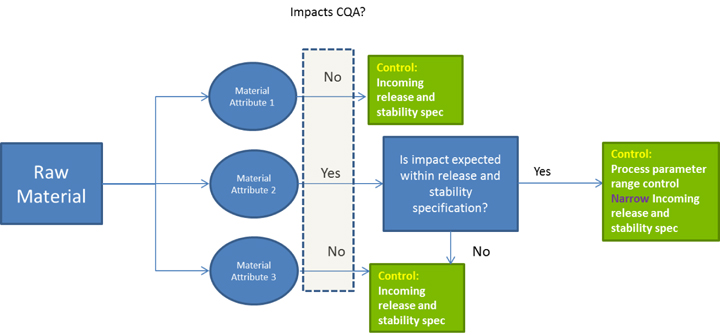
You will need to show that you understand the effect of raw material variability on your product as well as on your manufacturing process. Use of multiple lots during development can provide data on raw material lot-to-lot variability and its related effects on process and product. When that is not feasible, a manufacturer may consider including different lots of raw materials during bench-scale studies. In addition to the raw materials themselves, you should gain an understanding of whether and how raw material degradants might affect your process or product.
A QbD approach can use relevant knowledge to help you define how to go about setting specifications, in-process controls (IPCs), and handling conditions. Testing of Raw Materials The forum discussed what levels of testing are important for specific raw materials. A supplier’s certificate of analysis (CoA) is never sufficient for raw materials because good manufacturing practices (GMPs) require appropriate testing, and at a minimum, testing for identity. The material ordered may include additives, preservatives, degradation products, or contaminants. You must verify that the CoA is appropriate for control of the raw material, but you can’t assume that at the outset.
Similarly, CoA verification may be necessary only once a year once your experience with a given supplier has shown that quality is consistent. Vendor qualification is an important factor in defining your testing needs. To ensure the quality of raw materials against adulteration, identity testing is essential. Currently, tests with fingerprint techniques — e.g., nuclear magnetic resonance (NMR) imaging and Raman, nearinfrared (NIR), and Fourier-transform infrared (FTIR) spectroscopy — are used to assure the identity and quality of raw materials.

Whatever techniques you use, it is important to retain samples for future investigations. Photographic libraries of materials and their packaging have also proven useful for identifying and preventing use of counterfeit products. How often and in how much depth you need to verify a CoA through independent testing is an important consideration, especially for environments in which counterfeiting or contamination can occur.
Once you understand the CMAs of your raw materials, you need to identify which tests are relevant for testing specific quality attributes (QAs) of those raw materials. Sampling plans need careful consideration and should be risk based, dependent on the nature and use of the RM, and any regulatory requirements. Such plans should always be justified in a report available for inspection and/or filing.
It is important to consider RM stability and whether any special tests for degradants are needed for release of the material over time. A stability profile will dictate the purchasing program (storage of large quantities or buying as needed) as well as affect the associated testing strategy.
Supply Quality Management:
Ensuring Quality and Availability It is becoming increasingly evident in the current supply chain environment that management of suppliers and the “cold chain” is essential to assuring the quality of raw materials. How often and how thoroughly you perform vendor audits depends on your experience with a given vendor.
A manufacturer’s general experience with a vendor (prior knowledge) is an important criterion used to evaluate that vendor’s suitability to supply raw materials. Items to consider when selecting a vendor include its quality systems and its solvency, as well as its length of time in business, its geographic area, and whether it supplies multiple industries or just one or two drug manufacturers. Those form part of a risk assessment relating to suppliers to be described in more detail below.
Ensuring both the availability and qualification of secondary suppliers is important as well. Practices such as split purchasing may help ensure that you have good working relationships with multiple vendors. Strict change control sections should be included in supplier agreements and should include details requiring a vendor to notify you of changes in its product or suppliers. Such agreements should also provide for impact assessments from both supplier and manufacturer in the event that a supplier makes any changes. Supply chain traceability is not as straightforward as it might seem.
Although most manufacturers use country-of-origin (COO) questionnaires, those often prove less than ideal in revealing what you need to know. It is critical to craft questions that get the in-depth answers you need. For example, rather than asking “Do you purchase supplies from any high-risk countries?” you might ask “From what countries do you purchase supplies?” If the specified countries include any you consider to be high risk, you can follow up or choose another supplier.
It is critical to use risk-assessment techniques for determining traceability to avoid a false sense of security that can lead to costly or even deadly errors. It is sometimes unclear exactly what roles are played by whom in a supply chain.
Which companies are manufacturers, which are distributors, and which are intermediaries is not necessarily clear. A company that simply repackages a raw material from 55-gallon drums into smaller containers may consider itself a manufacturer. Due diligence will help ensure that you really know where your raw materials originated. As part of assessing supply chain complexity, forum participants were informed of a proposed program whereby industry creates a system of cooperative audits in which vendors would be audited by a selected team representing all industry rather than multiple auditors from each company continuously auditing suppliers.
The resulting audits would lead to certification that would assure all purchasers that each vendor meets certain defined criteria. Such a “360° Rx” program would enable increased depth of supplier audits and save manufacturers time and money (see box, right). The Role of Compendial Standards: Compendia provide some assurance of minimum quality standards for specified materials. However, compendial standards may differ among the pharmacopoeias.
Few of the complex raw materials (e.g., culture media, soy, yeastolates, most growth factors) used in biotechnology manufacturing are compendial, and those that are (e.g., insulin) may not have the appropriate compendial limits on specific quality attributes — or even test for quality attributes necessary to control pharmaceutical manufacturing. Even for standard chemical raw materials (e.g., trace metals), compendial standards may not focus on quality attributes relevant for biotechnology process and product quality assurance.
Those may be product- and/or process-specific. Furthermore, compendial standards do not necessarily help control for contamination, counterfeiting, or supply chain issues because a supplier can simply state it meets compendia — a statement that currently requires no certification
Risk Management
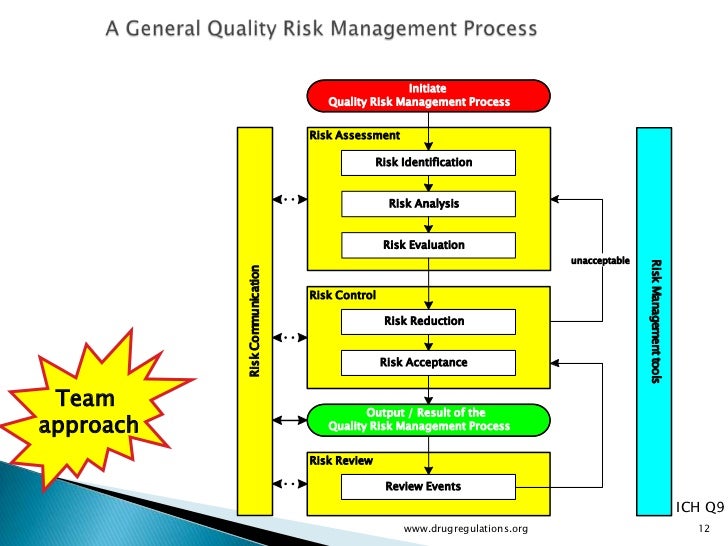
Risk assessments are an important tool for ensuring the safety, efficacy, consistency, and supply of pharmaceutical products. Many companies in both the United States and the European Union are using ICH Q9 as a basis for risk assessment, control, communication, and future review.
Risk assessments should begin by identifying all raw materials and assessing their criticality to product safety, efficacy, and supply. RM risk assessments require cross-functional input from all departments including supply, product development, manufacturing, quality control, quality assurance, clinical, and any other contributors. It was clear from this forum’s discussions that risk assessments are only as good as the people who carry them out. Having the right expertise over a spectrum of areas is vital if any risk assessment is to be meaningful. Multiple risk assessment tools exist, but in general, a good risk assessment must address concepts such as impact/ severity and likelihood/detectability.
One tool discussed at the forum (Figure 1) used nine blocks to score a supplier’s performance against material risk levels for audits, supplier qualification, supplier monitoring, change control, material specifications and testing, quality agreements, supplier certification, and sourcing, or other appropriate combinations of factors. Risk assessment should also be performed in relation to country of origin. It is critical to be able to trace your raw materials to their source. Just as a biopharmaceutical manufacturer audits its suppliers, those suppliers must also know, audit, and qualify their own distributors.
It is now well known that there are high-risk geographic areas where additional caution should be exercised to assure purity and identity of sourced materials. A potentially overlooked risk assessment issue is that manufacturers need to evaluate their raw materials and products in relation to opportunities for someone to make a profit through adulteration (e.g., by diluting a product to increase volume, and thus sales income). Any materials identified in such an evaluation should be managed with particular caution.

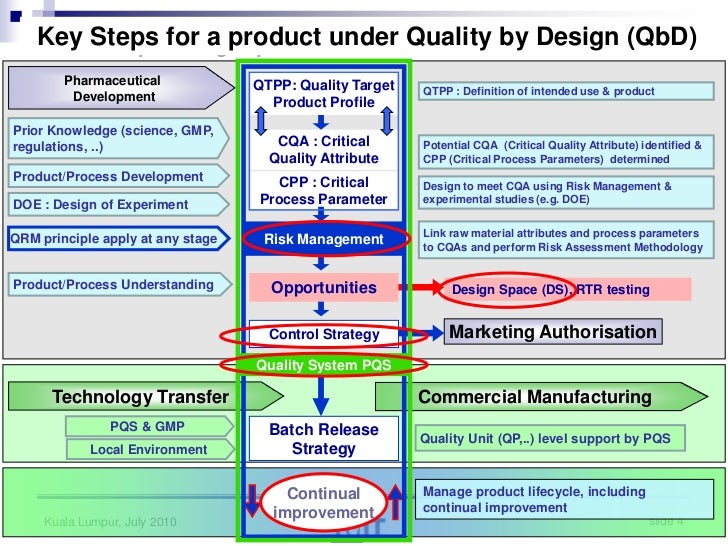
Risk assessments ensure that appropriate control strategies and raw materials (e.g., grade, origin) have been selected, which is relevant to a QbD approach. For regulatory filings, acceptable specifications, raw materials, and control strategies are tested with the necessary acceptance criteriia to ensure the performance of a process and the quality of its ultimate products. A periodic risk review should include more than a mere cursory review of individual risk assessments. It should reevaluate the risk program itself based on experience and lessons learned. Your risk assessment should be phase-appropriate, and as such it will change as data become available throughout development.
Early on, your raw materials risk assessment can be based on platform and previous knowledge, on the quality assurance of your suppliers, and adventitious agent introduction. As a manufacturing process develops, you will need to reevaluate that risk assessment including commercial considerations of scale and production frequency, highrisk raw materials control strategy, and handling and storage requirements.
During commercialization, design of experiments (DoEs) and collated knowledge will further define the CQAs of both product and RMs as well as potential and actual interactions among RMs, process, and product. At that point, you will be able to define and justify the raw materials for your commercial process and refine their specifications.
By the time your product is ready for market launch, you will have updated the failure modes and effects analysis (FMEA), completed your raw materials specifications, set your sourcing strategy, put in place your supplier qualification program, defined your raw material control strategy, and made your risk assessment ready for filing. The morning’s session resulted in a list of elements to be included in a raw materials risk assessment
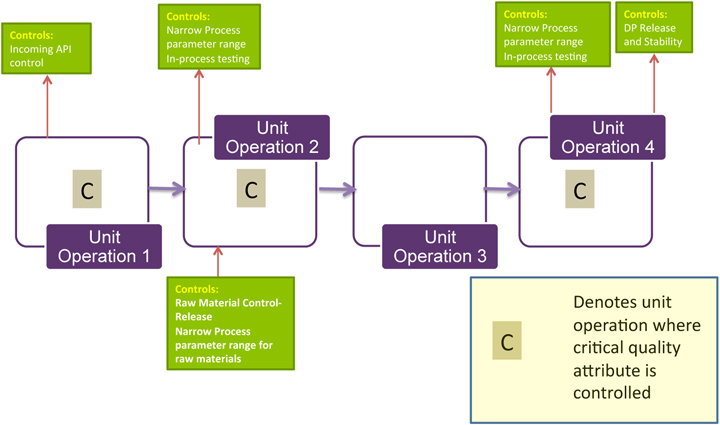
Elements of Raw Material
Risk Assessments Is the raw material biological, chemical, or physical (such as tubing or stoppers, materials that are not actual components of the end product)? How likely is the raw material to introduce biological or chemical contamination?
Is the raw material or are its degradants able to directly affect the safety and/or efficacy of a drug substance (e.g., toxicity, chemical modifications)?
How complex is the raw material itself or its impurity profile? How much prior knowledge (e.g., historical or published knowledge, current experience) do you have regarding the raw material? What is the Intended use of the raw material (e.g., as a buffer, reagent, or excipient)?
Where in the manufacturing process will this raw material will be used (upstream/ downstream)?
What is the extent of supply chain traceability (considering country of origin, supply chain complexity, and supply chain security)?
What is the extent of supplier quality assurance (from audits, monitoring, historical experience)?
How extensive is the characterization of the raw material (how well can the raw material be characterized; standard existing methods or novel techniques; the RM’s impact on test methods)?
How stable is the raw material? Is the raw material new to the process or a result of a change to an existing raw material (if a change, what studies have been executed to assure comparability)?
What is the depth of knowledge of the RM’s own manufacturing process to assess the risk associated with its use (e.g., process contaminants)?
Does the use of the raw material in a manufacturing environment present safety and/or handling risks? Does your process have the ability to clear the raw material?
Are there associated business risks (e.g., a solesource or multiple-source material, unique or not to the pharmaceutical industry, custom-made or not, and the supplier’s ability to consistently meet specific requirements)?
What is your level of understanding of the raw material CMAs?

Benefits of Implementing QbD
| Benefits for the FDAEnhances scientific foundation for review Provides for better coordination across review, compliance, and inspection Improves information in regulatory submissions Provides for better consistency Improves quality of review (establishing a quality management system for CMC) Provides for more flexibility in decision making Ensures decisions made on scientific and not on empirical information Involves various disciplines in decision making Uses resources to address higher risks |
Benefits for Industry Ensures better design of products with fewer problems in manufacturing Reduces number of manufacturing supplements required for postmarket changes; relies on process and risk understanding and risk mitigation Allows for implementation of new technology to improve manufacturing without regulatory scrutiny Allows for possible reduction in overall costs of manufacturing; creates less waste Ensures less hassle during review, reduces deficiencies, speeds approvals Improves interaction with the FDA; operates on a scientific rather than on a process level Allows for continuous improvements in products and manufacturing processes Allows for better understanding of how APIs and excipients affect manufacturing Relates manufacturing to clinical during design Provides a better overall business model |
Frequently Used QbD Terms
| Quality Attribute: A physical, chemical, or microbiological property or characteristic of a material that directly or indirectly alters quality | Critical Quality Attribute (CQA): A quality attribute that must be controlled within predefined limits to ensure that a product meets its intended safety, efficacy, stability, and performance |
| Real-Time Release (RTR): Ability to evaluate and ensure acceptable quality of an in-process and/or final product based on process data, including valid combination of assessment of material attributes by direct and/or indirect process measurements and assessment of critical process parameters and their effects on in-process material attributes | Process Parameter: An input variable or condition of a manufacturing process that can be directly controlled in the process. Typically, such parameters are physical or chemical (e.g., temperature, process time, column flow rate, column volume, reagent concentration, or buffer pH). |
| Critical Process Parameter (CPP): A process parameter whose variability has an influence on a CQA and therefore should be monitored or controlled to ensure a process produces a desired quality. | Process Performance Attribute: An output variable or outcome that cannot be directly controlled but is an indicator that a process performed as expected |
| Key Process Parameter (KPP): An input process parameter that should be carefully controlled within a narrow range and is essential for process performance; a key process parameter does not affect product quality attributes. If the acceptable range is exceeded, it may affect the process (e.g., yield, duration) but not product quality. | Non-Key Process Parameter: An input parameter that has been demonstrated to be easily controlled or has a wide acceptable limit. Such parameters may influence quality or process performance if acceptable limits are exceeded. |
| Design Space: The multidimensional combination and interaction of input variables (e.g., material attributes) and process parameters that have been demonstrated to provide assurance of quality; working within a design space is not considered to be a change requiring regulatory approval. Movement out of a design space is considered to be a change and would normally initiate a regulatory postapproval change process. Design space is proposed by an applicant and is subject to regulatory assessment and approval (ICH Q8). | Control Strategy: A planned set of controls, derived from current product and process understanding, that ensures process performance and product quality; such controls can include parameters and attributes related to drug substance and drug product materials and components, facility and equipment operating conditions, in-process controls, finished-product specifications, and associated methods, and frequency of monitoring and control (ICH Q10). |
| Quality Target Product Profile (QTPP): A prospective summary of the quality characteristics of a drug product that ideally will be achieved to ensure desired quality, taking into account safety and efficacy of a drug product |

2 Responses to “Raw Material Variation into QbD Risk Assessment”
Sorry, the comment form is closed at this time.
thanks
Good information. Thank you.