











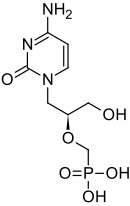
CIDOFOVIR
(S)-1-(3-Hydroxy-2-phosphonylmethoxypropyl)cytosine
[(S)-2-(4-Amino-2-oxo-1,2-dihydropyrimidin-2-yl)-1-(hydroxymethyl)ethoxymethyl]phosphonic acid
113852-37-2 CAS
120362-37-0 (Na salt)
149394-66-1 (dihydrate)
launched 1996 Gilead
SYNTHESIS.. CHEMDRUG
Rega Instituut (Originator)
For the treatment of CMV retinitis in patients with acquired immunodeficiency syndrome (AIDS)
US5142051 PATENT
| Canada | 1340856 | 1999-12-21 | EXPIRY 2016-12-21 |
| United States | 5142051 | 1993-06-26 | 2010-06-26 |
Cidofovir is a DNA polymerase inhibitor that was launched in 1996 by Gilead for the intravenous treatment of cytomegaloviral (CMV) retinitis in AIDS patients. Early clinical trials are underway at the National Institute for Allergy & Infectious Disease (NIAID) for the treatment of BK virus nephropathy (BKVN) in patients who have undergone kidney transplants.
Cidofovir suppresses CMV replication by selective inhibition of viral DNA synthesis. Biochemical data support selective inhibition of CMV DNA polymerase by cidofovir diphosphate, the active intracellular metabolite of cidofovir. Cidofovir diphosphate inhibits herpesvirus polymerases at concentrations that are 8- to 600-fold lower than those needed to inhibit human cellular DNA polymerases alpha, beta, and gamma1, 2, 3. Incorporation of cidofovir into the growing viral DNA chain results in reductions in the rate of viral DNA synthesis.
Cidofovir was originally developed under a collaboration between the Academy of Sciences of the Czech Republic and the Rega Institute for Medical Research. In 1991 and 1992, Gilead entered into license agreements with the Rega Institute that covered a large number of nucleotide analogue compounds and structures, including cidofovir. The drug became the subject of a marketing collaboration between Gilead and Pfizer (formerly Pharmacia & Upjohn) in August 1996 that covers all countries outside the U.S.
Cidofovir (brand name Vistide) is an injectable antiviral medication primarily used as a the treatment for cytomegalovirus (CMV) retinitis (an infection of the retina of the eye) in patients with AIDS.[1][2]
Its only indication that has received regulatory approval worldwide is cytomegalovirus retinitis.[1][2] Cidofovir has also shown efficacy in the treatment ofaciclovir-resistant HSV infections.[3] Cidofovir has also been investigated as a treatment for progressive multifocal leukoencephalopathy with successful case reports of its use.[4] Despite this meta-analyses have failed to demonstrate any efficacy in AIDS patients,[5] and the limited data in non-AIDS patients fail to demonstrate any efficacy either.[6] Cidofovir might have anti-smallpox efficacy and might be used on a limited basis in the event of a bioterror incident involving smallpox cases.[7] A cidofovir derivative with much higher activity against smallpox that can be taken orally has been developed.[8] It has inhibitory effects on varicella-zoster virus replication in vitro although no clinical trials have been done to date, likely due to the abundance of safer alternatives such as aciclovir.[9] Cidofovir shows anti-BK virus activity in a subgroup of transplant patients.[10] Cidofovir is being investigated as a complementary intralesional therapy against papillomatosis caused by HPV.[11][12]
It first received FDA approval on the 26th of June 1996,[13] TGA approval on the 30th of April 1998[2] and EMA approval on the 23rd of April 1997.[14]
Other
It has been suggested as an antitumour agent, due to its suppression of FGF2.[15][16]
Cidofovir was discovered at the Institute of Organic Chemistry and Biochemistry, Prague, by Antonín Holý, and developed by Gilead Sciences[20] and is marketed with the brand name Vistide by Gilead in the USA, and by Pfizerelsewhere.
The chemical name of cidofovir is 1-[(S)-3-hydroxy-2-(phosphonomethoxy)propyl]cytosine dihydrate (HPMPC), with the molecular formula of C8H14N3O6P•2H2O and a molecular weight of 315.22 (279.19 for anhydrous). The chemical structure is:

Cidofovir is a white crystalline powder with an aqueous solubility of ≥ 170 mg/mL at pH 6 to 8 and a log P (octanol/aqueous buffer, pH 7.1) value of -3.3.
Cidofovir Injection is a sterile, hypertonic aqueous solution for intravenous infusion only. The solution is clear and colorless. It is supplied in clear glass vials, each containing 375 mg of anhydrous cidofovir in 5 mL aqueous solution at a concentration of 75 mg/mL.
The formulation is pH-adjusted to 7.4 (range 7.1 to 7.7) with sodium hydroxide and/or hydrochloric acid and contains no preservatives. The appropriate volume of Cidofovir Injection must be removed from the single-use vial and diluted prior to administration
INTRODUCTION
Cidofovir’s chemical formula is C8H14N3O6P and its IUPAC name is ({[(S)-1-(4-amino-2-oxo-1,2-dihydropyrimidin-1-yl)-3-hydroxypropan-2-yl]oxy}methyl)phosphonic acid. Cidofovir has also been described as (S)-(1-(4-amino-2-oxopyrimidin-1(2H)-yl)-3-hydroxypropan-2-yloxy)methylphosphonic acid as well as possibly by other chemical names. Its chemical structure is:

Cidofovir was discovered at the Institute of Organic Chemistry and Biochemistry, Prague, and developed by Gilead Sciences. Today, cidofovir is an injectable antiviral medication for the treatment of cytomegalovirus (CMV) retinitis in patients with AIDS. It suppresses CMV replication by selective inhibition of viral DNA polymerase and therefore prevention of viral replication and transcription. It is an acyclic nucleoside phosphonate, and is therefore independent of phosphorylation by viral enzyme, in contrast to, for instance, acyclovir.
Cidofovir is marketed with the brand name Vistide® by Gilead in the United States and by Pfizer in other parts of the world. Vistide® is a sterile, hypertonic aqueous solution for intravenous infusion only. The solution is clear and colorless. It is supplied in clear glass vials, each containing 375 mg of anhydrous cidofovir in 5 mL aqueous solution at a concentration of 75 mg/mL. The formulation is pH-adjusted to 7.4 with sodium hydroxide and/or hydrochloric acid and contains no preservatives. Renal impairment is the major toxicity of Vistide®.
Presently, there are no Orange Book patents listed as having claims which cover Vistide®, although previously U.S. Pat. No. 5,142,051 was listed in the Orange Book for Vistide®. The ‘051 patent is not directed specifically to cidofovir or its crystalline forms. Instead, it broadly discloses N-phosphonylmethoxyalkyl derivatives of pyrimidine and purine bases.
Cytomegalovirus (Cytomegaoviyns, CMV) is one of the biggest dangers of the herpes virus, the body’s infection rates as high as 50% to 80% of the current adult prevalence rate of more than 95%, generally showed a latent infection, most infections had no clinical symptoms, but under certain conditions, the invasion of organs and systems to produce more severe disease. The virus can invade the lung, liver, kidney, salivary gland, mammary gland and other polymorphonuclear leukocytes and lymphocytes, and, since the long-term or intermittent saliva, milk sweat, blood, urine, semen, exclude uterine secretions of the virus. Spread through a variety of ways in the mouth, genital tract, placenta, blood transfusion or organ transplantation.
When the body’s immune dysfunction, such as infected with HIV, cancer patients undergoing radiotherapy, chemotherapy, organ or bone marrow transplantation immunosuppressive anti-rejection etc will stimulate active infection, can cause acute retinitis, interstitial pneumonia, gastroenteritis and encephalitis, blindness or death without treatment rate of over 70%. With the rise in HIV infection rates and organ transplants extensively for anti-CMV drugs is also increasing demand.
cidofovir (cidofovir, HPMPC) are novel ether derivatives of cytidine phosphono chemical name
[5]-NL [(3 – hydroxy-2 – methoxy-phosphonic acid) glycerol]-N4-cytosine, Molecular structure of the formula (I):

Gilead developed by the United States, in May 1996 the FDA approved injectable celecoxib Duofu Wei listed, France and Canada also continued with the approval of the use of the trade name Vistide. Its CAS number is 113852-37-2, formula C8H14N3O6P, the structure of formula (I). Cidofovir for CMV is highly inhibitory activity of certain ganciclovir or foscarnet resistant strains of the virus are also active. And herpes simplex virus (HSV), herpes zoster virus (VZV), human papillomavirus (HPV), also has a strong activity.
Its mechanism of action: cidofovir having a phosphoric acid group, a ring-opening mechanism of the antiviral nucleoside phosphonate compound (ANP) and the consistent cyclic nucleoside analogues are nucleosides or virus in vivo kinase activation into triphosphate metabolite, thereby inhibiting viral replication by DNA polymerase and reverse transcriptase. Unlike the three-step cyclic nucleoside analogues must phosphorylation reaction, ring opening nucleoside phosphonate group containing phosphorus compound itself, eliminating the first step of the phosphorylation reaction speed, and thus a higher activity. Cidofovir is absorbed when the cells in the cell pyrimidine nucleoside phosphorylase kinase (P bandit kinase and NDP kinase) to effect conversion of the active metabolite monophosphate (HPMPCp), diphosphate (HPMPCpp) and a bile acid base adducts. Cidofovir diphosphate inhibits viral DNA polymerase or reverse transcriptase activity, and its corresponding natural dNTP incorporated into the viral DNA chain competition, since no 3 – hydroxy end, continue to extend the DNA chain termination. Can slow the synthesis of DNA, viral DNA and to the loss of stability, thereby inhibiting viral replication, transcription of the ability to reduce viral DNA to exert antiviral activity. Compared with other anti-CMV drugs, cidofovir characteristics: significant and lasting effect, started the first two weeks administered once a week, then only administered once every two weeks, easy to use, and to reduce its toxicity side effects.
Several major techniques are based on the synthesis of cidofovir cytosine as starting material, mainly carried out to improve the synthesis of the side chain.
(I) J. Med Chem, 1989,32,1457 ~ 1463 discloses a synthetic process:

The route to cytosine as the raw material, with a chiral side chain by condensation, deprotection and reduction can be obtained in three steps cidofovir.However, chiral side chain subject to a six-step reaction system. The total yield is low, adverse side. And using Me3SiBr, so that the costs and the risk of surge, is not conducive to industrial production.
(2) US 5591852,1995-1-7; US 2005/023833 & WO 2006/014429 and US 2009/0270618, Tetrahedron Lett 1994,35,3243-3246 and “Chinese Journal of New Drugs”, 2007,16. , 1272-1274 for the synthesis of a lot of improvements:

Benzoyl cytosine with a chiral starting material and trityloxymethyl ethylene oxide condensation, deprotection and hydrolysis was then prepared by deprotection cidofovir group. The synthetic steps to make some shorter, but still use expensive Me3SiBr, adverse ones, the low yield of the security at the cost of industrial production is still unfavorable. (Several different patent protection only in the order of the amino cytosine different!)
(3) Patent Publication No. CN1690065A, CN1690066A, CN1690067A (2005 年 11 月 2 Publication Date) and the “Chinese Journal of Medicinal Chemistry” 2007,17,41-46, reported a new synthetic route:

The route of process steps is too long, the total yield is low, side effects side. But not conducive to industrial production.
(4) Patent No. CN 101205215A (25 June 2008 publicly) announced a halogen epoxy propane as a starting material for the synthesis route:
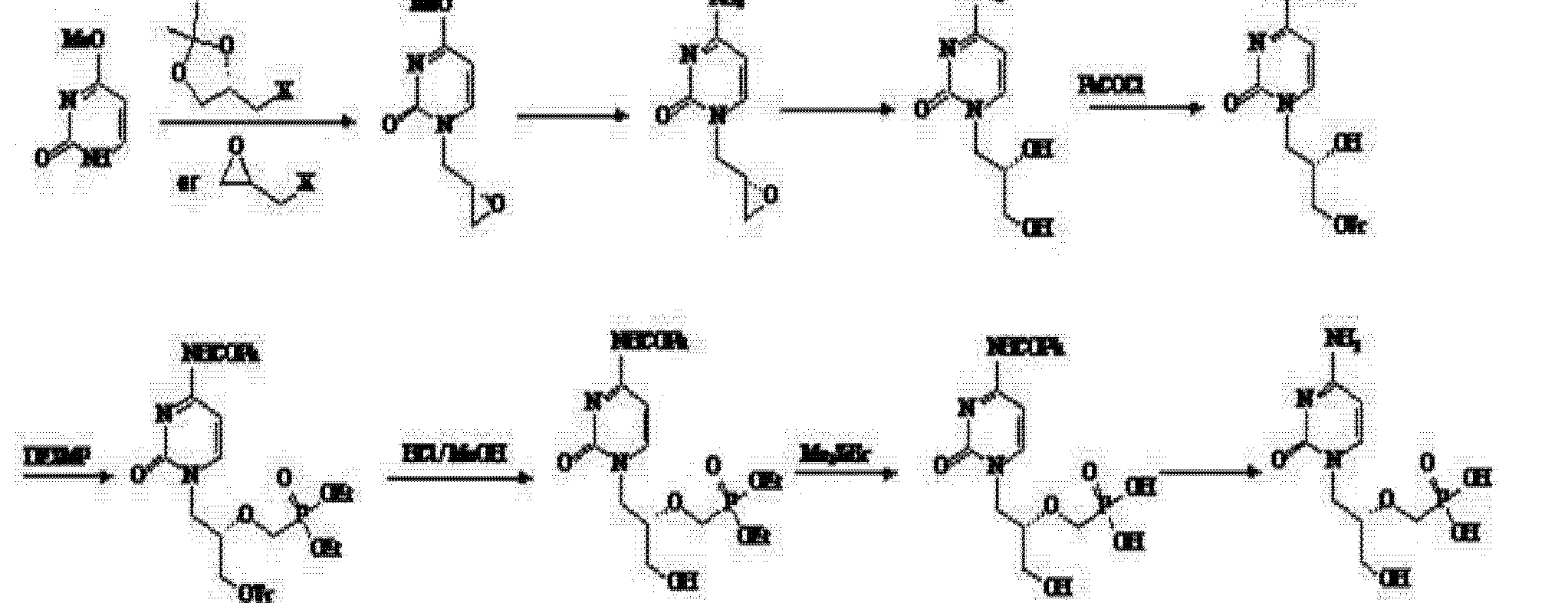
Use of the route (R) – epihalohydrin reaction with cytosine, cytosine ring because alkaline easily cause epoxy ring-opening reaction of the ring, but side reactions, the purified product is not, nor is suitable for industrial production.
Subsequently, the patent number CN 101525352A (2009 年 9 月 9 Publication Date) discloses (4) based on the modified route through epoxypropionate alkane ether in the form of a direct reaction with cytosine, after a series of similar steps obtain the final product cidofovir.
In view of the clinical application of cidofovir more favorable therapeutic effect in, looking for a high yield and because of economic and practical, easy to control, the risk of small synthetic methods and technology is now more urgent needs.
Synthesis

Brodfuehrer, P; Howell, Henry G.; Sapino, Chester; Vemishetti, Purushotham (1994). “A practical synthesis of (S)-HPMPC”. Tetrahedron Letters 35 (20): 3243. doi:10.1016/S0040-4039(00)76875-4.
………………………………………
CN 102268040

, Example 1:
1 Synthesis of 4,4 ‘- dimethoxytrityl methyl – (R) – glycidol (Compound III): The 5 04 g (15 mmoDDMT-Cl grain port 0 20 g (1 52 mmol… ) 4_ dimethylaminopyridine (DMAP) was dissolved in 100 mL CH2C12 cooled to 0 ° C, was added dropwise 10 mL TEA was slowly added 2. 00 g (27mmol) hydroxymethyl chiral oxirane (Compound II ) addition was completed, the reaction warmed to room temperature naturally. fly 4 h, until TLC until the disappearance of the detection DMT-Cl, the reaction was stopped by filtration, the filtrate was washed with saturated NaHC03 solution (50mLX2), saturated NaCl solution (50 mLX2), anhydrous Na2S04 dried, filtered, and concentrated to a viscous colorless directly, i.e., 5 08 g of 4,4 ‘-dimethoxy-triphenylmethyl _ -.. (R) – glycidol (Compound III), yield 90 %, HPLC purity 99%.
2, Synthesis (S)-N1_ [(2 – hydroxy-3 – (dimethoxytrityl) propyl] cytosine (Compound IV):. Under nitrogen to 3 56 g (32 mmol) of cytosine was added 150 mL of anhydrous N, N-dimethylformamide (DMF), and at room temperature, was added portionwise 1. 24 g (31 mmol, molar concentration of 60%) NaH, 0. 5 h after adding 11 92 g (31 mmol) 4,4 ‘-. dimethoxytrityl methyl – (R) – glycidol (Compound III), plus finished warming up to 10 (Tll (TC reaction . 6-8 h and then filtered, and the filtrate evaporated under reduced pressure DMF, the remaining solid phase was added 500 mL of ethyl acetate and 50 mL of water, separated and the organic layer was washed with saturated NaHC03 solution (50 mL X 2), saturated NaCl solution (50 mL X 2), dried over anhydrous Na2S04 filtered and dried, and concentrated to give 13 90 g of a white solid, S Jie (S)-Nl-[(2 -.. hydroxy-3 – (methoxy-dimethoxytrityl ) propyl] cytosine (Compound IV), yield 92%, HPLC purity 98%.
3 Synthesis ⑶-Nl-{[2_ (phosphonic acid methoxy diethoxy) -3 – (methoxy-dimethoxytrityl)] propyl} cytosine (Compound V):
75 ~ 80 ° C under the conditions, 48 76 g (0 100 mol.) (S) _N1_ [(2 – hydroxy-3 – (dimethoxytrityl) propyl]. Cytosine (Compound IV) was added to 150 mL anhydrous DMF, and then inputs 8. 5g (0. 050 mol) tert-butoxide, magnesium reaction 0.5-1 h, tosyloxy added diethyl 32 methylsulfinyl . 2 g (0. 100 mol), the reaction epileptic 8 h, p-toluenesulfonic acid was added to neutralize the excess alkali to neutral distilled DMF, ethyl acetate (300 mLX 3) washing the combined ethyl acetate phase was concentrated to give a solid, i.e., synthetic 58 18 g (S)-Nl-. {[2 – (diethoxy-phosphono-methoxy) -3 – (methoxy-dimethoxytrityl)] propyl} cytosine (Compound V), yield 89%, HPLC purity greater than 95%.
4 Synthesis of (S)-Nl-{[2_ (phosphonic acid methoxy diethoxy) -3 – hydroxy] propyl} cytosine (Compound VI): The 10 g (S)-Nl- {[2 – (phosphono-methoxy ethoxy) -3 – (methoxy-dimethoxytrityl)] propyl}-cell
Pyrimidine (compound V) was dissolved in a concentration of 70 mL of 80% acetic acid solution, 90 ° C reaction. After 5 h, cooled to room temperature, 50 mL of water and 30 mL of dichloromethane, and the organic phase washed with water (30 mL X2) and the combined aqueous phase was concentrated to give crude 9. 5 g, can be performed directly in the next reaction.
can also be separated by flash column chromatography (CH2C12 = MeOH = 10: 1), 4.6 g obtained as a pale yellow oil, i.e. (S)-Nl-{[2 – (methoxy diethoxy phosphono ) -3 – hydroxy] propyl} cytosine (Compound VI), yield 90%.
5 was synthesized ⑶-Nl-{[2_ (diphosphonic acid methoxy) -3 – hydroxy] propyl} cytosine (Compound I):
The 9.5g (S)-Nl-{[2 – (methoxy diethoxy phosphonomethyl) -3 – hydroxy] propyl} cytosine (Compound VI) into a crude product containing 5 76 g (0.. 045 mol) solution of hydrogen iodide, hydroiodic acid, and after reflux for 4-5 h. (50 mLX 2) wash solution was separated with ethyl acetate. The aqueous phase was added sodium hydroxide to adjust pH between 3 Γ3 6, filtered, recrystallized from methanol to give 3.81 g of white crystalline solid, S Jie (S)-Ni-{[2 -.. (Diphosphonic acid methoxy yl) -3 – hydroxy] propyl} cytosine (Compound I), yield 88% (containing two crystal water), HPLC purity greater than 99%.
…………………………………………
POLYMORPHS
Example 7 Amorphous Cidofovir
Intermediate 5 (FIG. 7; 0.5 g, 0.054 mol) was heated with a solution of sodium methoxide in methanol (0.5 M, 15 mL, 7.5 mmol) at 72° C. for 14.5 h then at 90° C. for 5.5 h. The reaction mixture was quenched with water (10 mL) and filtered through a bed of ion exchange resin Dowex® 50WX8 100-200 (H). The filtrate was cycled through the ion exchange bed (2 times) then washed successively with 1:1 methanol:water (40 mL), methanol (40 mL) and 4% triethylamine:methanol (50 mL). This ion-exchange bed was further washed with 48:48:4 methanol:water:triethylamine (100 mL) until no UV absorbance was detected in the filtrate. This reaction produced intermediate 7 (FIG. 7) together with cyclic cidofovir impurity. This mixture was then dissolved in 6 N HCl and heated to 65° C. After cooling the reaction mixture to room temperature, ethyl acetate was charged and stirred and the aqueous layer separated. The aqueous was stirred with ethanol (50 mL). The precipitated material was filtered and the solid was washed with ethanol. The ethanol filtrate was concentrated. The concentrated material was taken up in acetonitrile and stirred with trimethylsilyl bromide (19 mL) at room temperature for 18 h. The reaction mixture was filtered and the filtrate concentrated. The residue was taken up in toluene (30 mL) and ammonium hydroxide (28%, 50 mL) was charged and stirred at room temperature. The organic phase was separated and the aqueous phase was concentrated to dryness. Water (20 mL) and ethanol (15 mL) were added to the residue. The mixture pH was 6 and was adjusted to pH 3 with concentrated HCl (2 mL) then adjusted to pH 4 to 4.5 with 28% NH4OH. After stirring for 0.5 h, the mixture was cooled, filtered and the solids washed with 2:1 EtOH:H2O and dried under vacuum for 18 h. The isolated solid was taken up in water (10 mL) and 28% NH4OH added to give a solution. Concentrated HCl was added to the solution until pH 4 was reached. Ethanol (13 mL) was charged and the mixture stirred at −17° C. for 18 h, filtered and the solids washed with 2:1 EtOH:water (2×8 mL), dried under vacuum at 35° C. The cidofovir isolated in this manner was determined to be in the amorphous form by XRPD.

……………………………..
Journal of the American Chemical Society, 2011 , vol. 133, 7 p. 2264 – 2274
http://pubs.acs.org/doi/abs/10.1021/ja109823e

………………………………………………..
READ ALSO
Synthesis and antiviral activity of the nucleotide analogue (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine
J Med Chem 1989, 32(7): 1457
http://pubs.acs.org/doi/abs/10.1021/jm00127a010
References
- “Vistide (cidofovir) dosing, indications, interactions, adverse effects, and more”.Medscape Reference. WebMD. Retrieved 4 February 2014.
- “Product Information VISTIDE®”. TGA eBusiness Services. Gilead Sciences Pty Ltd. 3 September 2013. Retrieved 5 February 2014.
- Chilukuri, S; Rosen, T (2003 Apr). “Management of acyclovir-resistant herpes simplex virus.”.Dermatologic clinics 21 (2): 311–20. doi:10.1016/S0733-8635(02)00093-1.PMID 12757254.
- Segarra-Newnham M, Vodolo KM (June 2001). “Use of cidofovir in progressive multifocal leukoencephalopathy”. Ann Pharmacother 35 (6): 741–4. doi:10.1345/aph.10338.PMID 11408993.
- De Luca, A; Ammassari, A; Pezzotti, P; Cinque, P; Gasnault, J; Berenguer, J; Di Giambenedetto, S; Cingolani, A; Taoufik, Y; Miralles, P; Marra, CM; Antinori, A; Gesida 9/99, IRINA, ACTG 363 Study, Groups (September 2008). “Cidofovir in addition to antiretroviral treatment is not effective for AIDS-associated progressive multifocal leukoencephalopathy: a multicohort analysis.”. AIDS (London, England) 22 (14): 1759–67.doi:10.1097/QAD.0b013e32830a5043. PMID 18753934.
- Langer-Gould, A; Atlas, SW; Green, AJ; Bollen, AW; Pelletier, D (28 July 2005). “Progressive Multifocal Leukoencephalopathy in a Patient Treated with Natalizumab”. New England Journal of Medicine 353 (4): 375–381. doi:10.1056/NEJMoa051847. PMID 15947078.
- De Clercq E (July 2002). “Cidofovir in the treatment of poxvirus infections”. Antiviral Res. 55(1): 1–13. doi:10.1016/S0166-3542(02)00008-6. PMID 12076747.
- Bradbury, J (March 2002). “Orally available cidofovir derivative active against smallpox.”.Lancet 359 (9311): 1041. doi:10.1016/S0140-6736(02)08115-1. PMID 11937193.
- Magee, WC; Hostetler, KY; Evans, DH (August 2005). “Mechanism of Inhibition of Vaccinia Virus DNA Polymerase by Cidofovir Diphosphate”. Antimicrobial Agents and Chemotherapy49 (8): 3153–3162. doi:10.1128/AAC.49.8.3153-3162.2005. PMC 1196213.PMID 16048917.
- Araya CE, Lew JF, Fennell RS, Neiberger RE, Dharnidharka VR (February 2006).“Intermediate-dose cidofovir without probenecid in the treatment of BK virus allograft nephropathy”. Pediatr Transplant 10 (1): 32–7. doi:10.1111/j.1399-3046.2005.00391.x.PMID 16499584.
- Broekema FI, Dikkers FG (August 2008). “Side-effects of cidofovir in the treatment of recurrent respiratory papillomatosis”. Eur Arch Otorhinolaryngol 265 (8): 871–9. doi:10.1007/s00405-008-0658-0. PMC 2441494. PMID 18458927.
- Soma MA, Albert DM (February 2008). “Cidofovir: to use or not to use?”. Curr Opin Otolaryngol Head Neck Surg 16 (1): 86–90. doi:10.1097/MOO.0b013e3282f43408.PMID 18197029.
- “Cidofovir Monograph for Professionals – Drugs.com”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 5 February 2014.
- “Vistide : EPAR -Product Information” (PDF). European Medicines Agency. Gilead Sciences International Ltd. 7 November 2013. Retrieved 5 February 2014.
- Liekens S, Gijsbers S, Vanstreels E, Daelemans D, De Clercq E, Hatse S (March 2007). “The nucleotide analog cidofovir suppresses basic fibroblast growth factor (FGF2) expression and signaling and induces apoptosis in FGF2-overexpressing endothelial cells”. Mol. Pharmacol.71 (3): 695–703. doi:10.1124/mol.106.026559. PMID 17158200.
- Liekens S (2008). “Regulation of cancer progression by inhibition of angiogenesis and induction of apoptosis”. Verh. K. Acad. Geneeskd. Belg. 70 (3): 175–91. PMID 18669159.
- Rossi, S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3. edit
- “Vistide (cidofovir)” (package insert). Gilead Sciences. September 2010. DOSAGE AND ADMINISTRATION: Dosage.
- Safrin, S; Cherrington, J; Jaffe, HS (September 1997). “Clinical uses of cidofovir”. Reviews in Medical Virology 7 (3): 145–156. doi:10.1002/(SICI)1099-1654(199709)7:3<145::AID-RMV196>3.0.CO;2-0. PMID 10398479.
- “Press Releases: Gilead”. Retrieved 2007-12-05.
- Synthesis and antiviral activity of the nucleotide analogue (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine
J Med Chem 1989, 32(7): 1457 - Synthesis and antiherpesvirus activity of (S)-1-((3-hydroxy-2-phosphonylmethoxy)propyl)cytosine (HPMPC) and related nucleotide analoguesNucleosides Nucleotides 1989, 8(5-6): 923
- Journal of Pharmaceutical Sciences, 2012 , vol. 101, 9 p. 3249 – 326
- BRODFUEHRER P R ET AL: “A Practical Synthesis of (S)-HPMPC“, 19940101, vol. 35, no. 20, 1 January 1994 (1994-01-01), pages 3243-3246, XP002012084
- http://www.chemdrug.com/databases/8_0_nhnpseknoegbdisx.html CHEMDRUG SYNTHESIS
| 2 | * | BRONSON, JOANNE J. ET AL: “Synthesis and antiviral activity of nucleotide analogs bearing the (S)-(3-hydroxy-2-phosphonylmethoxy)propyl moiety attached to adenine, guanine, and cytosine“, ACS SYMPOSIUM SERIES , 401(NUCLEOTIDE ANALOGUES ANTIVIRAL AGENTS), 88-102 CODEN: ACSMC8; ISSN: 0097-6156, 1989, XP002677024, |
| 3 | * | BRONSON, JOANNE J. ET AL: “Synthesis and antiviral activity of the nucleotide analog (S)-1-[3-hydroxy-2-(phosphonylmethoxy)prop yl]cystosine“, JOURNAL OF MEDICINAL CHEMISTRY , 32(7), 1457-63 CODEN: JMCMAR; ISSN: 0022-2623, 1989, XP002677026, |
| 4 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; JIANG, XING-KAI ET AL: “Practical synthesis of a broad-spectrum antiviral drug cidofovir”, XP002677148, retrieved from STN Database accession no. 2009:1568897 -& JIANG XING-KAI ET AL: “Practical synthesis of a broad-spectrum antiviral drug cidofovir“, JIEFANGJUN YAOXUE XUEBAO – PHARMACEUTICAL JOURNAL OF CHINESE PEOPLE’S LIBERATION ARMY, ZHONGGUO RENMIN JIEFANGJUN, ZONGHOUQINBU, WEISHENGBU, YAOPIN YIQI JIANYANSUO, CN, vol. 25, no. 5, 1 January 2009 (2009-01-01), pages 395-397, XP008152443, ISSN: 1008-9926 |
| 5 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; LIU, JIANFENG ET AL: “Improved synthesis of cidofovir”, XP002677149, retrieved from STN Database accession no. 2008:1052727 -& LIU JIANFENG ET AL: “Improved synthesis of cidofovir“, ZHONGGUO YAOWU HUAXUE ZAZHI – CHINESE JOURNAL OF MEDICINAL CHEMISTRY, GAI-KAI BIANJIBU, SHENYANG, CN, vol. 17, no. 1, 1 February 2007 (2007-02-01), pages 41-43, XP008152444, ISSN: 1005-0108 |
| 6 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; LU, NING ET AL: “Synthesis of cidofovir”, XP002677151, retrieved from STN Database accession no. 2007:1124844 -& LU NING ET AL: “Synthesis of cidofovir“, ZHONGGUO XIN YAO ZAZHI – CHINESE NEW DRUGS JOURNAL, GAI-KAN BIANJIBU, BEIJING, CN, vol. 16, no. 16, 1 January 2007 (2007-01-01), pages 1272-1274, XP008152436, ISSN: 1003-3734 |
| 7 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; YI, HONG ET AL: “Synthesis of antiviral agent cidofovir”, XP002677150, retrieved from STN Database accession no. 2007:1249882 -& YI HONG ET AL: “Synthesis of antiviral agent cidofovir“, ZHONGGUO KANGSHENGSU ZAZHI/ CHINESE JOURNAL OF ANTIBIOTICS, SICHUAN, CN, vol. 31, no. 7, 1 January 2006 (2006-01-01), pages 412-413, XP008152413, ISSN: 1001-8689 |
| 8 | * | FROMTLING R A ET AL: “Cidofovir. HPMPC. GS-504. GS-0504. Viside“, DRUGS OF THE FUTURE, PROUS SCIENCE, ES, vol. 21, no. 10, 1 January 1996 (1996-01-01), pages 1003-1013, XP008152410, ISSN: 0377-8282 |
| 9 | * | HOLY A: “SYNTHESES OF ENANTIOMERIC N-(3-HYDROXY-2-PHOSPHONOMETHOXYPROPYL) DERIVATIVES OF PURINE AND PYRIMIDINE BASES“, COLLECTION OF CZECHOSLOVAK CHEMICAL COMMUNICATIONS, INSTITUTE OF ORGANIC CHEMISTRY & BIOCHEMISTRY, PRAGUE; CZ, vol. 58, no. 3, 1 January 1993 (1993-01-01), pages 649-674, XP009042514, ISSN: 0010-0765, DOI: 10.1135/CCCC19930649 |
| 10 | * | JOANNE J BRONSONA ET AL: “A New Synthesis of the Potent and Selective Anti-Herpesvirus Agent (S)-1-[3-Hydroxy-2-(Phosphonylmethoxy)Prop yl]Cytosine“, NUCLEOSIDES, NUCLEOTIDES AND NUCLEIC ACIDS, TAYLOR & FRANCIS, PHILADELPHIA, PA , vol. 9, no. 6 1 January 1990 (1990-01-01), pages 745-769, XP008152412, ISSN: 1525-7770, DOI: 10.1080/15257779008043142 Retrieved from the Internet: URL:http://www.tandfonline.com/doi/abs/10.1080/15257779008043142 [retrieved on 2006-10-04] |
| 11 | * | PETR ALEXANDER AND ANTONÍN HOL: “General Method of Preparation of N-[(S)-(3-Hydroxy-2-phosphonomethoxypropyl )] Derivatives of Heterocyclic Bases“, COLLECTION OF CZECHOSLOVAK CHEMICAL COMMUNICATIONS, INSTITUTE OF ORGANIC CHEMISTRY & BIOCHEMISTRY, PRAGUE; CZ, vol. 58, no. 5, 1 May 1993 (1993-05-01), pages 1151-1163, XP008152404, ISSN: 0010-0765, DOI: 10.1135/CCCC19931151 |
| 12 | * | SNOECK, ROBERT ET AL: “(S)-1-(3-Hydroxy-2-phosphonylmethoxypropy l)cytosine, a potent and selective inhibitor of human cytomegalovirus replication“, ANTIMICROBIAL AGENTS AND CHEMOTHERAPY , 32(12), 1839-44 CODEN: AMACCQ; ISSN: 0066-4804, 1988, XP002677023, |
| 13 | * | SYMERSK AND A HOL J: “Structure of 1-(S)-(3-hydroxy-2-phosphonylmethoxypropyl )cytosine; an antiviral agent“, ACTA CRYSTALLOGRAPHICA SECTION C. CRYSTAL STRUCTURE COMMUNICATIONS, MUNKSGAARD, COPENHAGEN, DK, vol. 47, no. 10, 15 October 1991 (1991-10-15), pages 2104-2107, XP008152403, ISSN: 0108-2701, DOI: 10.1107/S0108270190013257 |
| 14 | * | ULRIKA ERIKSSON: “Synthesis and biological evaluation of novel cidofovir prodrugs targeting HPepT1“, SYNTHESIS AND BIOLOGICAL EVALUATION OF NOVEL CIDOFOVIR PRODRUGS TARGETING HPEPT1- A DISSERTATION PRESENTED TO THE FACULTY OF THE GRADUATE SCHOOL UNIVERSITY OF SOUTHERN CALIFORNIA IN PARTIAL FULFILLMEN , 1 January 2005 (2005-01-01), pages 1-287, XP008152406, Retrieved from the Internet: URL:http://search.proquest.com/pqdtscieng/docview/305421788/fulltextPDF/1371C222DB27F96BE2D/2?accountid=29404 |
| 15 | * | WEBB, ROBERT R., II ET AL: “Synthesis of (S)-N1-(3-hydroxy-2-phosphonylmethoxy)prop ylcytosine, (S)-HPMPC“, TETRAHEDRON LETTERS , 29(43), 5475-8 CODEN: TELEAY; ISSN: 0040-4039, 1988, XP002677025 |
| CN1559429A * | Feb 18, 2004 | Jan 5, 2005 | 肖广常 | 注射用西多福韦冻干粉针剂 |
| CN1690066B * | Apr 19, 2004 | Apr 28, 2010 | 横店集团成都分子实验室有限公 | 抗病毒剂西多福韦新衍生物 |
| CN101525352A * | Feb 16, 2009 | Sep 9, 2009 | 苏州凯达生物医药技术有限公司 | 西多福韦及其中间体的制备方法 |
| CN102268040A * | Sep 5, 2011 | Dec 7, 2011 | 扬州三友合成化工有限公司 | 抗病毒药物西多福韦的一种合成方法 |
| US5142051 | Jul 17, 1987 | Aug 25, 1992 | Ceskoslovenska Akademie Ved | N-phosphonylmethoxyalkyl derivatives of pyrimidine and purine bases and a therapeutical composition therefrom with antiviral activity |






 300
300 

















 ……………………………………..Vinita Gupta, Chief Executive Officer, Lupin Limited;
……………………………………..Vinita Gupta, Chief Executive Officer, Lupin Limited;














 PEMETREXED
PEMETREXED













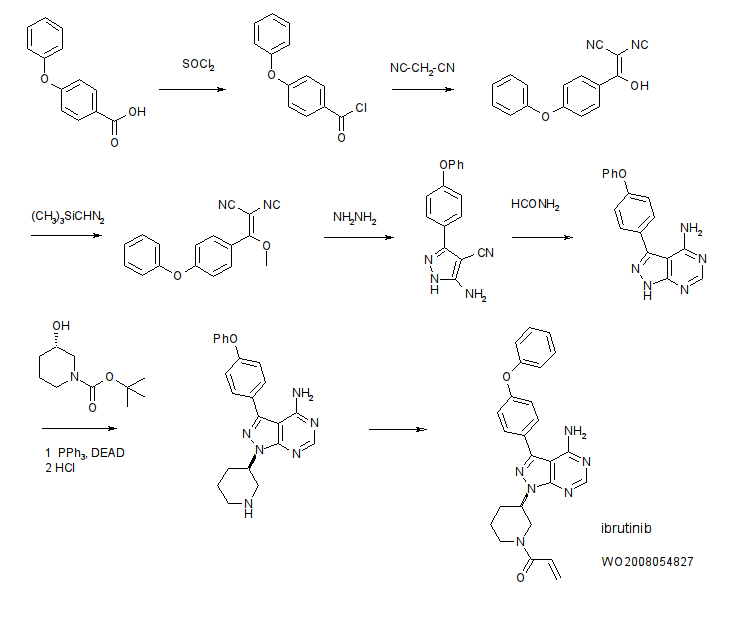
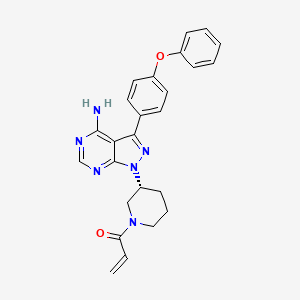 IBRUTINIB
IBRUTINIB




















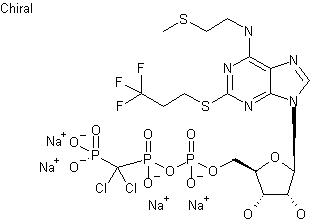
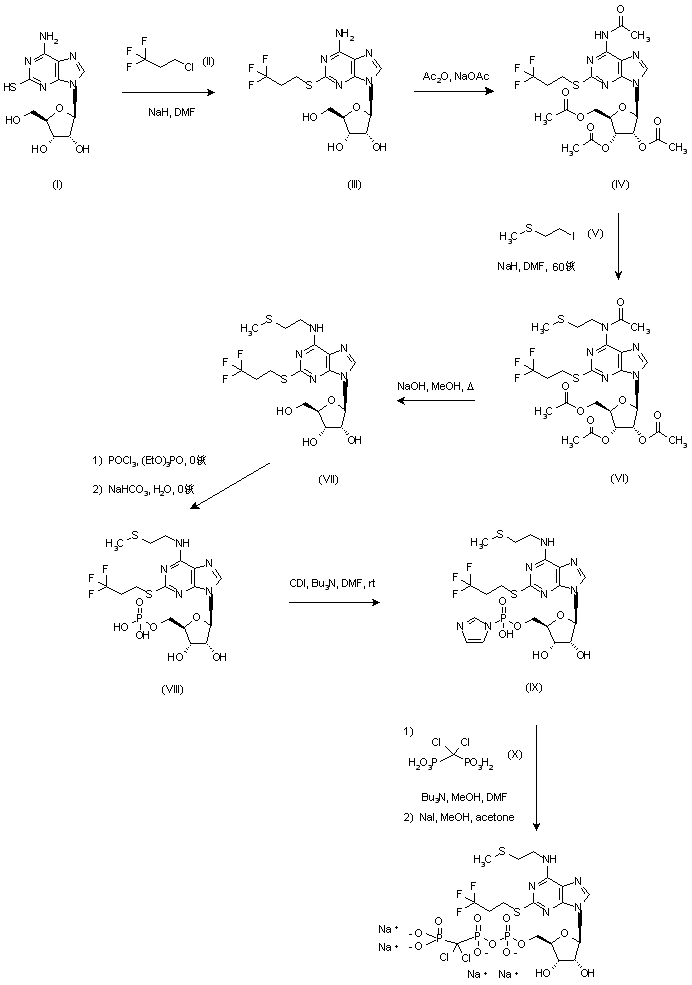
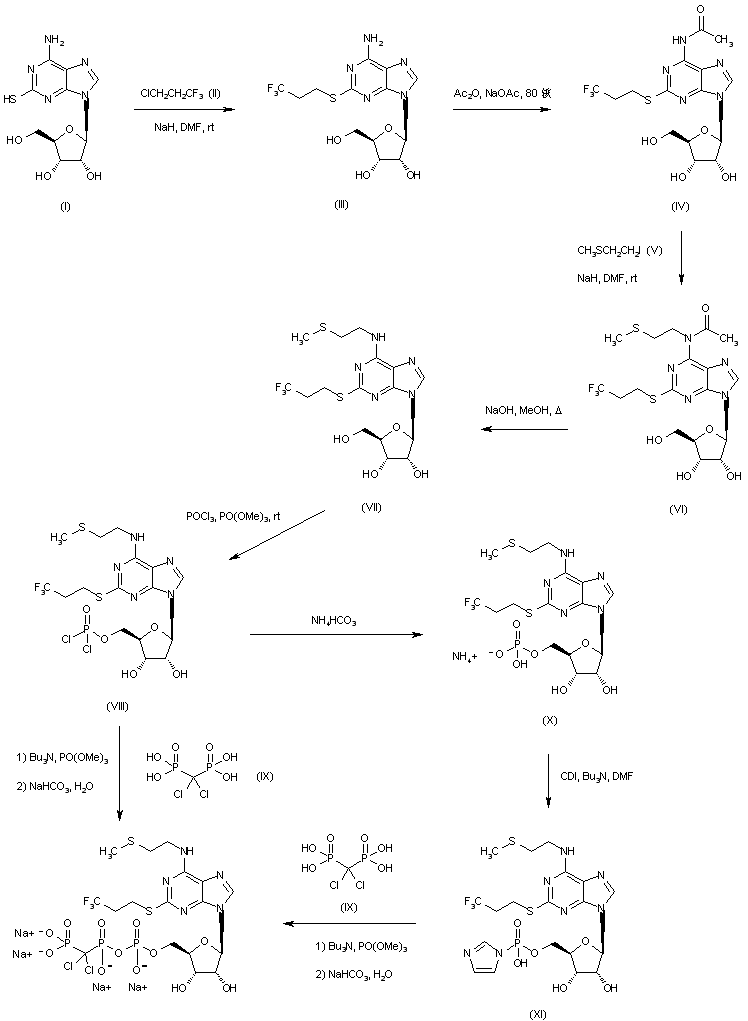
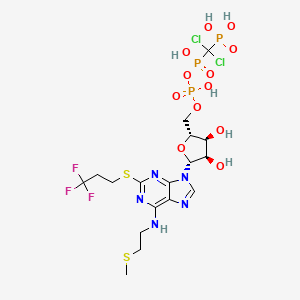 Cangrelor
Cangrelor


 OR
OR

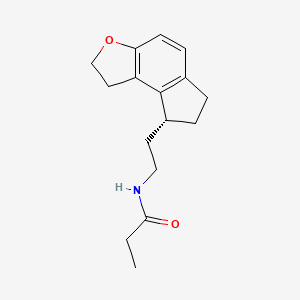 Ramelteon
Ramelteon



































 RAMELTEON
RAMELTEON




















 SONIDEGIB
SONIDEGIB