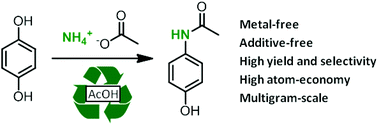
Selinexor (KPT-330)
1393477-72-9
Karyopharm Therapeutics, Inc.
WO2011109799A1
WO2013019548A1
Synonyms
-
(Z)-3-(3-(3,5-Bis(trifluoromethyl)phenyl)-1H-1,2,4-triazol-1-yl)-N’-(pyrazin-2-yl)acrylohydrazide
-
2-Propenoic acid, 3-(3-(3,5-bis(trifluoromethyl)phenyl)-1H-1,2,4-triazol-1-yl)-, 2-(2-pyrazinyl)hydrazide, (2Z)-
- 3-[3-[3,5-Bis(trifluoromethyl)phenyl]-1H-1,2,4-triazol-1-yl]-N’-(pyrazin-2-yl)acrylohydrazide
Karyopharm Announces Initiation of Phase 2 Study of Selinexor (KPT-330) in Patients with …
MarketWatch
“These patients were treated in our Phase 1 clinical trial of Selinexor in … Additional Phase 1 and Phase 2 studies are ongoing or currently planned and … the discovery and development of novel first-in-class drugs directed against …

Selinexor, a Exportin-1 (CRM1/XPO1) agonist, is in phase II clinical trials at Karyopharm for the treatment of advanced or metastatic gynecological malignancies (cervical, ovarian and uterine carcinomas) and recurrent glioblastomas. The company is also evaluating the compound in early clinical trials for the treatment of advanced solid tumors, hematological cancer (non-Hodgkin’s lymphoma, multiple myeloma and Waldenstrom’s macroglobulinemia), soft tissue or bone sarcoma, relapsed or refractory acute myeloid leukemia (AML) and relapsed or refractory acute lymphoblastic leukemia (ALL).
In 2014, orphan drug designation was assigned in U.S. for the treatment of acute myeloid leukemia and diffuse large B-cell lymphoma
Cells from most major human solid and hematologic malignancies exhibit abnormal cellular localization of a variety of oncogenic proteins, tumor suppressor proteins, and cell cycle regulators (Cronshaw et al. 2004, Falini et al 2006). For example, certain p53 mutations lead to localization in the cytoplasm rather than in the nucleus. This results in the loss of normal growth regulation, despite intact tumor suppressor function. In other tumors, wild-type p53 is sequestered in the cytoplasm or rapidly degraded, again leading to loss of its suppressor function. Restoration of appropriate nuclear localization of functional p53 protein can normalize some properties of neoplastic cells (Cai et al. 2008; Hoshino et al. 2008; Lain et al. 1999a; Lain et al. 1999b; Smart et al. 1999), can restore sensitivity of cancer cells to DNA damaging agents (Cai et al. 2008), and can lead to regression of established tumors (Sharpless & DePinho 2007, Xue et al. 2007). Similar data have been obtained for other tumor suppressor proteins such as forkhead (Turner and Sullivan 2008) and c-Abl (Vignari and Wang 2001). In addition, abnormal localization of several tumor suppressor and growth regulatory proteins may be involved in the pathogenesis of autoimmune diseases (Davis 2007, Nakahara 2009). CRMl inhibition may provide particularly interesting utility in familial cancer syndromes (e.g. , Li-Fraumeni Syndrome due to loss of one p53 allele,
BRCA1 or 2 cancer syndromes), where specific tumor suppressor proteins (TSP) are deleted or dysfunctional and where increasing TSP levels by systemic (or local) administration of CRMl inhibitors could help restore normal tumor suppressor function. Specific proteins and R As are carried into and out of the nucleus by specialized transport molecules, which are classified as importins if they transport molecules into the nucleus, and exportins if they transport molecules out of the nucleus (Terry et al. 2007;
Sorokin et al. 2007). Proteins that are transported into or out of the nucleus contain nuclear import/localization (NLS) or export (NES) sequences that allow them to interact with the relevant transporters. Chromosomal Region Maintenance 1 (Crml or CRM1), which is also called exportin-1 or Xpol, is a major exportin.
Overexpression of Crml has been reported in several tumors, including human ovarian cancer (Noske et al. 2008), cervical cancer (van der Watt et al. 2009), pancreatic cancer (Huang et al. 2009), hepatocellular carcinoma (Pascale et al. 2005) and osteosarcoma (Yao et al. 2009) and is independently correlated with poor clinical outcomes in these tumor types.
Inhibition of Crml blocks the exodus of tumor suppressor proteins and/or growth regulators such as p53, c-Abl, p21, p27, pRB, BRCA1, IkB, ICp27, E2F4, KLF5, YAP1, ZAP, KLF5, HDAC4, HDAC5 or forkhead proteins (e.g., FOX03a) from the nucleus that are associated with gene expression, cell proliferation, angiogenesis and epigenetics. Crml inhibitors have been shown to induce apoptosis in cancer cells even in the presence of activating oncogenic or growth stimulating signals, while sparing normal (untransformed) cells. Most studies of Crml inhibition have utilized the natural product Crml inhibitor Leptomycin B (LMB). LMB itself is highly toxic to neoplastic cells, but poorly tolerated with marked gastrointestinal toxicity in animals (Roberts et al. 1986) and humans (Newlands et al. 1996). Derivatization of LMB to improve drug-like properties leads to compounds that retain antitumor activity and are better tolerated in animal tumor models (Yang et al. 2007, Yang et al. 2008, Mutka et al. 2009). Therefore, nuclear export inhibitors could have beneficial effects in neoplastic and other proliferative disorders.
In addition to tumor suppressor proteins, Crml also exports several key proteins that are involved in many inflammatory processes. These include IkB, NF-kB, Cox-2, RXRa, Commdl, HIFl, HMGBl, FOXO, FOXP and others. The nuclear factor kappa B (NF-kB/rel) family of transcriptional activators, named for the discovery that it drives immunoglobulin kappa gene expression, regulate the mRNA expression of variety of genes involved in inflammation, proliferation, immunity and cell survival. Under basal conditions, a protein inhibitor of NF-kB, called IkB, binds to NF-kB in the nucleus and the complex IkB-NF-kB renders the NF-kB transcriptional function inactive. In response to inflammatory stimuli, IkB dissociates from the IkB-NF-kB complex, which releases NF-kB and unmasks its potent transcriptional activity. Many signals that activate NF-kB do so by targeting IkB for proteolysis (phosphorylation of IkB renders it “marked” for ubiquitination and then proteolysis). The nuclear IkBa-NF-kB complex can be exported to the cytoplasm by Crml where it dissociates and NF-kB can be reactivated. Ubiquitinated IkB may also dissociate from the NF-kB complex, restoring NF-kB transcriptional activity. Inhibition of Crml induced export in human neutrophils and macrophage like cells (U937) by LMB not only results in accumulation of transcriptionally inactive, nuclear IkBa-NF-kB complex but also prevents the initial activation of NF-kB even upon cell stimulation (Ghosh 2008, Huang 2000). In a different study, treatment with LMB inhibited IL-Ιβ induced NF-kB DNA binding (the first step in NF-kB transcriptional activation), IL-8 expression and intercellular adhesion molecule expression in pulmonary microvascular endothelial cells (Walsh 2008). COMMDl is another nuclear inhibitor of both NF-kB and hypoxia-inducible factor 1 (HIFl) transcriptional activity. Blocking the nuclear export of COMMDl by inhibiting Crml results in increased inhibition of NF-kB and HIFl transcriptional activity (Muller 2009).
Crml also mediates retinoid X receptor a (RXRa) transport. RXRa is highly expressed in the liver and plays a central role in regulating bile acid, cholesterol, fatty acid, steroid and xenobiotic metabolism and homeostasis. During liver inflammation, nuclear RXRa levels are significantly reduced, mainly due to inflammation-mediated nuclear export of RXRa by Crml . LMB is able to prevent IL-Ιβ induced cytoplasmic increase in RXRa levels in human liver derived cells (Zimmerman 2006).
The role of Crml -mediated nuclear export in NF-kB, HIF-1 and RXRa signalling suggests that blocking nuclear export can be potentially beneficial in many inflammatory processes across multiple tissues and organs including the vasculature (vasculitis, arteritis, polymyalgia rheumatic, atherosclerosis), dermatologic (see below), rheumatologic
(rheumatoid and related arthritis, psoriatic arthritis, spondyloarthropathies, crystal arthropathies, systemic lupus erythematosus, mixed connective tissue disease, myositis syndromes, dermatomyositis, inclusion body myositis, undifferentiated connective tissue disease, Sjogren’s syndrome, scleroderma and overlap syndromes, etc.).
CRM1 inhibition affects gene expression by inhibiting/activating a series of transcription factors like ICp27, E2F4, KLF5, YAP1, and ZAP.
Crml inhibition has potential therapeutic effects across many dermatologic syndromes including inflammatory dermatoses (atopy, allergic dermatitis, chemical dermatitis, psoriasis), sun-damage (ultraviolet (UV) damage), and infections. CRMl inhibition, best studied with LMB, showed minimal effects on normal keratinocytes, and exerted anti-inflammatory activity on keratinocytes subjected to UV, TNFa, or other inflammatory stimuli (Kobayashi & Shinkai 2005, Kannan & Jaiswal 2006). Crml inhibition also upregulates NRF2 (nuclear factor erythroid-related factor 2) activity, which protects keratinocytes (Schafer et al. 2010, Kannan & Jaiswal 2006) and other cell types (Wang et al. 2009) from oxidative damage. LMB induces apoptosis in keratinocytes infected with oncogenic human papillomavirus (HPV) strains such as HPV 16, but not in uninfected keratinocytes (Jolly et al. 2009).
Crml also mediates the transport of key neuroprotectant proteins that may be useful in neurodegenerative diseases including Parkinson’s disease (PD), Alzheimer’s disease, and amyotrophic lateral sclerosis (ALS). For example, by (1) forcing nuclear retention of key neuroprotective regulators such as NRF2 (Wang 2009), FOXA2 (Kittappa et al. 2007), parking in neuronal cells, and/or (2) inhibiting NFKB transcriptional activity by sequestering IKB to the nucleus in glial cells, Crml inhibition could slow or prevent neuronal cell death found in these disorders. There is also evidence linking abnormal glial cell proliferation to abnormalities in CRMl levels or CRMl function (Shen 2008).
Intact nuclear export, primarily mediated through CRMl, is also required for the intact maturation of many viruses. Viruses where nuclear export, and/or CRMl itself, has been implicated in their lifecycle include human immunodeficiency virus (HIV), adenovirus, simian retrovirus type 1, Borna disease virus, influenza (usual strains as well as H1N1 and avian H5N1 strains), hepatitis B (HBV) and C (HCV) viruses, human papillomavirus (HPV), respiratory syncytial virus (RSV), Dungee, Severe Acute Respiratory Syndrome coronavirus, yellow fever virus, West Nile virus, herpes simplex virus (HSV), cytomegalovirus (CMV), and Merkel cell polyomavirus (MCV). (Bhuvanakantham 2010, Cohen 2010, Whittaker 1998). It is anticipated that additional viral infections reliant on intact nuclear export will be uncovered in the future.
The HIV-1 Rev protein, which traffics through nucleolus and shuttles between the nucleus and cytoplasm, facilitates export of unspliced and singly spliced HIV transcripts containing Rev Response Elements (RRE) RNA by the CRMl export pathway. Inhibition of Rev-mediated RNA transport using CRMl inhibitors such as LMBor PKF050-638 can arrest the HIV-1 transcriptional process, inhibit the production of new HIV-1 virions, and thereby reduce HIV-1 levels (Pollard 1998, Daelemans 2002). Dengue virus (DENV) is the causative agent of the common arthropod-borne viral disease, Dengue fever (DF), and its more severe and potentially deadly Dengue hemorrhagic fever (DHF). DHF appears to be the result of an over exuberant inflammatory response to DENV. NS5 is the largest and most conserved protein of DENV. CRMl regulates the transport of NS5 from the nucleus to the cytoplasm, where most of the NS5 functions are mediated. Inhibition of CRMl -mediated export of NS5 results in altered kinetics of virus production and reduces induction of the inflammatory chemokine interleukin-8 (IL-8), presenting a new avenue for the treatment of diseases caused by DENV and other medically important flaviviruses including hepatitis C virus (Rawlinson 2009).
Other virus-encoded RNA-binding proteins that use CRMl to exit the nucleus include the HSV type 1 tegument protein (VP 13/14, or hUL47), human CMV protein pp65, the SARS Coronavirus ORF 3b Protein, and the RSV matrix (M) protein (Williams 2008, Sanchez 2007, Freundt 2009, Ghildyal 2009).
Interestingly, many of these viruses are associated with specific types of human cancer including hepatocellular carcinoma (HCC) due to chronic HBV or HCV infection, cervical cancer due to HPV, and Merkel cell carcinoma associated with MCV. CRMl inhibitors could therefore have beneficial effects on both the viral infectious process as well as on the process of neoplastic transformation due to these viruses.
CRMl controls the nuclear localization and therefore activity of multiple DNA metabolizing enzymes including histone deacetylases (HDAC), histone acetyltransferases (HAT), and histone methyltransferases (HMT). Suppression of cardiomyocyte hypertrophy with irreversible CRMl inhibitors has been demonstrated and is believed to be linked to nuclear retention (and activation) of HDAC 5, an enzyme known to suppress a hypertrophic genetic program (Monovich et al. 2009). Thus, CRMl inhibition may have beneficial effects in hypertrophic syndromes, including certain forms of congestive heart failure and hypertrophic cardiomyopathies.
CRMl has also been linked to other disorders. Leber’s disorder, a hereditary disorder characterized by degeneration of retinal ganglion cells and visual loss, is associated with inaction of the CRMl switch (Gupta N 2008). There is also evidence linking
neurodegenerative disorders to abnormalities in nuclear transport.
…………………………………………
PATENT
http://www.google.com/patents/WO2013019548A1?cl=en
To date, however, small-molecule, drug-like Crml inhibitors for use in vitro and in vivo are uncommon. SUMMARY OF THE INVENTION
The present invention relates to compounds, or pharmaceutically acceptable salts thereof, useful as nuclear transport modulators. The invention also provides
pharmaceutically acceptable compositions comprising compounds of the present invention and methods of using said compounds and compositions in the treatment of various disorders, such as those associated with abnormal cellular responses triggered by improper nuclear transport..
In one embodiment of the invention, the compounds are represented by formula I:

http://www.google.com/patents/WO2013019548A1?cl=en
HERE IT REFERS AS 1-16 READER PLEASE CHECKABOVE AND BELOW FOR ERROR

HERE IT REFERS AS 1-18 READER PLEASE CHECK
http://www.google.com/patents/WO2013019548A1?cl=en
Example 1 : Synthesis of Intermediate (Z)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-lH-l,2,4- triazol-l-yl)acrylic acid.
Synthesis of 3,5-bis(trifluoromethyl)benzothioamid
A 2-L, 3-necked, round-bottomed flask was charged with a solution of 3,5- bis(trifluoromethyl)benzonitrile (200 g) in DMF (1 L). The solution was then treated with NaSH (123.7 g, 2.0 eq.) and MgCl2 (186.7 g, 1.0 eq.) and the reaction mixture was stirred at RT for 3 hours. The mixture was poured into an ice-water slurry (10 L) and the compound was extracted with EtOAc (3 x 1 L). The combined organic layers were washed with aqueous saturated brine (3 x 100 mL), dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure to afford 205 g of desired crude 3,5- bis(trifluoromethyl)benzothioamide (yield: 90 %), which wasused without purification in the following step.
Synthesi -(3,5-bis(trifluoromethyl)phenyl)-lH-l 2,4-triazole:
A 5-L, 3-necked, round-bottomed flask was charged with a solution of 3,5- bis(trifluoromethyl)benzothioamide (205.65 g) in DMF (1.03 L). Hydrazine hydrate (73.2 mL, 2.0 eq.) was added dropwise and the reaction mixture was stirred at RT for 1 h. HCOOH (1.03 L) was added dropwise and the reaction mixture was refluxed at 90 °C for 3 hours. After being allowed to cool to RT, the reaction mixture was poured into saturated aqueous sodium bicarbonate solution (7 L) and extracted with EtOAc (3 x 1 L). The combined organic layers were washed with aqueous saturated brine (3 x 500 mL), dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure (35 °C, 20 mmHg) to afford 180 g of crude compound. This crude material was stirred with petroleum ether (3 x 500 mL) , filtered and dried to obtain 160 g. of 3-(3,5-bis(trifluoromethyl)phenyl)-lH- 1,2,4-triazole obtained as a pale yellow solid (yield: 75%).
Synthesis of (Z)-isopropyl 3-(3-(3,5-bis(trifluoromethyl)phenyl)-lH-l,2,4-triazol-l- yl)acrylate:

A 2-L, 3-necked, round-bottomed flask was charged with a solution of 3-(3,5- bis(trifluoromethyl)phenyl)-lH-l ,2,4-triazole (160 g) in DMF (960 mL). The solution was treated with DABCO (127.74 g, 2 eq.) and stirred for 30 min before adding (Z)-isopropyl 3- iodoacrylate (150.32 g, 1.1 eq.) dropwise. After ca. 1 hour, the reaction mixture was poured into an ice-water slurry (5 L) and extracted with EtOAc (3 x 1 L). The combined organic layers were washed with aqueous saturated brine (3 x 100 mL), dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure (35 °C, 20 mmHg) to afford 250 g of crude compound that was purified by column chromatography (60/120 silica gel) using a ethyl acetate/n-hexane gradient (the column was packed in hexane and the desired compound started eluting from 2% EtOAC/n-hexane). Fractions containing the desired compounds were combined to afford 138 g the pure desired compound (yield: 61%).
Synthesis of (Z)-3 -(3 -(3 ,5-bis(trifluoromethyl)phenyl)- 1 H- 1 ,2,4-triazol- 1 -yl)acrylic acid:
In a 5-L, 3-necked, round-bottomed flask, (Z)-isopropyl 3-(3-(3,5- bis(trifluoromethyl)phenyl)-lH-l,2,4-triazol-l-yl)acrylate (130 g, 1.0 eq.) was dissolved in THF (1.3 L). A solution of LiOH (69.3 g, 5.0 eq.) in water (1.3 L) was added dropwise to the solution and the reaction mixture was stirred at room temperature for 4 h before being quenched with 400 mL ice-water slurry and made acidic (pH = 2-3) with dilute aqueous HC1. The mixture was extracted with EtOAc (3 x 1 L) and the combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure to afford 110 g of desired carboxylic acid (yield: 94 %) (cis content = 90.0%, trans content = 8.2% by LCMS).
Example 17: Synthesis of (E)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-lH-l,2,4-triazol-l-yl)- ‘-(pyrazin-2-yl)acrylohydrazide
Synthesis of 3,5-bis(trifluoromethyl)benzothioamide:
A 2-L, 3 -necked, round-bottomed flask, charged with a solution of 3,5- bis(trifluoromethyl)benzonitrile (200 g) in DMF (1 L), was treated with NaSH (123.7 g, 2.0 eq.) and MgCl2 (186.7 g, 1 eq.). The reaction mixture was stirred at RT for 3 h before being poured into an ice-water slurry (10 L) and was extracted with EtOAc (3 x 1 L). The combined organic extracts were washed with brine (3 x 100 niL), dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure (25 °C, 20 mmHg) to afford 205 g of crude compound (yield: 90 %), which was used in the following step without further purification.
Synthesis of 3-(3,5-bis(trifluoromethyl)phenyl)-lH-l,2,4-triazole:
A 5-L, 3-necked, round-bottomed flask, charged with a solution of 3,5- bis(trifluoromethyl)benzothioamide (205.65 g) in DMF (1.03 L) was treated with hydrazine hydrate (73.16 mL, 2.0 eq.) added dropwise. The reaction mixture was stirred at room temperature for 1 h before being treated with HCOOH (1.028 L) added dropwise. The reaction mixture was refluxed at 90°C for 3 h then cooled to room temperature and poured into saturated aqueous NaHC03 solution (7 L) and extracted with EtOAc (3 x 1L). The combined organic layers were washed with brine (3 x 500 mL), dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure (35°C, 20 mmHg) to afford 180 g of a solid. The solid was suspended in petroleum ether and the suspension was stirred, filtered and dried to afford the desired triazole as a pale yellow solid (160 g, yield: 75%).
Synthesis of (Z)-isopropyl 3-(3-(3,5-bis(trifluoromethyl)phenyl)-lH-l,2,4-triazol-l- yl)acrylate and (E)-isopropyl 3-(3-(3,5-bis(trifluoromethyl)phenyl)-lH-l,2,4-triazol-l- yl)acrylate:

A 2-L, 3-necked, round-bottomed flask, charged with a solution of 3-(3,5- bis(trifluoromethyl)phenyl)-lH-l,2,4-triazole (160 g,) in DMF (0.96 L, 6V), was treated with DAB CO (127.74 g, 2 eq.) and stirred for 30 min. (Z)-isopropyl 3-iodoacrylate (150.32 g, 1.1 eq.) was added dropwise to the above reaction mixture and stirred for 1 h before being poured into an ice-water slurry (5 L) and extracted with EtOAc (3 x 1 L). The combined organic extracts were washed with brine (3 x 100 mL), dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure (35°C, 20 mmHg) to afford 250 g of crude compound. Purification by column chromatography (Si02, 60/120 mesh, elution with EtOAc:hexanes gradient; the desired compounds started eluting in 2-2.5 % EtOAc in hexanes) afforded pure cis ester (138 g, yield: 61.6%) and pure trans ester (11.6 g, yield: 5.2%). Synthesis of (E)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-lH-l ,2,4-triazol-l-yl);
acid:
A 500-mL, 3 -necked, round-bottomed flask was charged with a solution of (E)- isopropyl 3-(3-(3,5-bis(trifluoromethyl)phenyl)-lH-l ,2,4-triazol-l-yl)acrylate (5.0 g) in THF (50 mL). The solution was treated with a solution of LiOH (2.66 g, 5.0 eq.) in water (50 mL) and the reaction mixture was stirred at room temperature for 4 h. before being diluted with 40 mL water, acidified (pH = 2-3) with dilute aqueous HC1 and extracted with EtOAc (3 x 100 mL). The organic extract was washed with brine, dried over anhydrous Na2S04, filtered and concentrated under reduced pressure to afford 2.75 g of the desired unsaturated carboxylic acid (yield: 61.6 %, purity: 99.0 % by LCMS).
Synthesis of (E)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-lH-l,2,4-triazol-l-yl)-N’- (pyrazin-2-yl)acrylohydrazide :

To a solution of (E)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-lH-l,2,4-triazol-l- yl)acrylic acid (0.75 g,) in EtOAc (25 mL) and THF (12.5 mL) was added a solution of 2- hydrazinopyrazine (0.23 g) in 12 mL THF at room temperature. T3P (50% in ethyl acetate, 1.52 mL) and DIPEA (1.46 mL) were added dropwise and simultaneously and the reaction mixture was stirred for 30 min at room temperature before being quenched with ice-cold water and extracted with EtOAc (3 x 25 mL). The combined organic layers were washed with brine, dried over anhydrous Na2S04 and concentrated under reduced pressure (35°C, 20 mmHg), affording 0.698 g of a crude solid. Trituration first with petroleum ether then with Et20 afforded 275 mg (yield: 29%) (E)-3-(3-(3,5-bis(trifiuoromethyl) phenyl)- 1H- 1,2,4- triazol-l-yl)-N’-(pyrazin-2-yl)acrylohydrazide. 1H NMR (400 MHz, DMSO-d6) δ ,10.3 (s, 1H), 9.15 (s, 2H), 8.59 (s, 2H), 8.30-8.26 (d, J= 14.8 Hz, 1H), 8.13 (s, 1H), 8.06-8.07 (m, 1H), 6.98-6.95 (d, J= 13.4 Hz, 1H); LCMS for Ci7H12F6N70 [M+H]+ 443.31 ; found 444.19 (RT 2.625 min, purity: 99.06%).
MY SUGESTION TO U
http://www.google.com/patents/WO2013019548A1?cl=en
(Z)-isopropyl 3-(3-(3,5-bis(trifluoromethyl)phenyl)-lH-l,2,4-triazol-l- yl)acrylate IS THE INTERMEDIATE
any discussion mail amcrasto@gmail.com
NOTE IF U USE Z OR CIS STARTING INTERMEDIATE U WILL GET Z ISOMER
…………………………………………………………
int 75 in
http://www.google.com/patents/WO2011109799A1?cl=en

Exam le 75
Molecular Weight: 239.12 Molecular Weight: 273.2 Molecular Weight: 281 .2
Molecular Weight: 393.3
[00715] Synthesis of Intermediate 1)
Molecular Weight: 239.12 Molecular Weight: 273.2
[00716] In a 100-mL, 3N round-bottomed flask equipped with nitrogen inlet, and a rubber septum, 3,5-bis(trifluoromethyl)benzonitrile (5.0 g,1.0 eq) dissolved in DMF (50 mL,10V),Added NaSH(3.09 g,2.0eq) and MgC12 (4.24 g,l eq).Reaction mixture was stirred at RT for 2-3h. The progress of reaction was followed by TLC analysis on silica gel with 40%EtOAc- hexane as mobile phase. SM Rf=0.5 and Product Rf=0.3. Reaction mixture was poured in to ice water (250mL) and extracted with EtOAc ( 3x 100 mL). The combined organic layers were washed with brine solution (3xl00mL), dried over MgS04, filtered, and concentrated by rotary evaporation (25°C, 20mmHg) to afford 5.0g of Crude compound which was used for next step without any purification, Yield (87.5%). Mass [M+l]+: 273.8
[00717] Synthesis of Intermediate-2
Molecular Weight: 273.20 Molecular Weight: 281 .16
[00718] In a 250-mL, 3N round-bottomed flask equipped with nitrogen inlet, and a rubber septum, Intermediate- 1(5.0 g, 1.0 eq.) was dissolved in DMF (50 mL,10V),added NH2NH2.H20 (25.0 mL,5V). The reaction mixture was stirred at RT for 1 h. To this reaction mixture HCOOH (25.0 mL, 5V) was added and reaction mixture was refluxed at 90 0 for 2-3 h. The progress of reaction was followed by TLC analysis on silica gel with 50% Ethyl acetate-n-Hexane as mobile phase. SM Rf=0.50 and Product Rf=0.3. Reaction mixture was poured into ice water (500 mL) and neutralized with saturated sodium bicarbonate solution. The reaction mixture was extracted with EtOAc (3×100 mL). The combined organic layers were washed with brine solution,(3xl00mL), dried over MgS04, filtered, and concentrated by rotary evaporation (25°C, 20mmHg) to afford 4.6g of crude compound, yield (89.49%). Mass: 279.6(-ve mode).
Molecular Weight: 281.2 Molecular Weight: 393.3
[00719] In a 100-mL, 3N round-bottomed flask equipped with nitrogen inlet, and a rubber septum, Intermediate-2(4.5 g, 1.0 eq.) was dissolved in DCM(45 mL,10V),added TEA (2.10 g, 1.3 eq) and isopropyl propiolate (2.33 g, 1.3 eq). The Reaction mixture was stirred at RT for 30 min. The progress of reaction was followed by TLC analysis on silica gel with 50% Ethyl acetate-Hexane as mobile phase, SM f=0.30 and Product Rf=0.5. Reaction mixture was concentrated by rotary evaporation (25°C, 20mmHg) to afford 5.8 g of Crude compound. The crude reaction mixture was purified by column chromatography using silica 60/120 using Ethyl acetate: Hexane as mobile phase. The column (5x10cm) was packed in Hexane and started eluting in Ethyl acetate in gradient manner starting with fraction collection(50-mL fractions) from 5 % to 20 % Ethyl acetate in hexane. Compound started eluting with 20% Ethyl acetate in Hexane. Fraction containing such TLC profile was collected together to obtain pure compound (1.4 g), Yield (22.26%).1H NMR: CDC13, 400 MHz) δ 9.74(s,lH),5 8.63(s,2H),5 7.95(s,lH),5 7.28-7.3 l(d,J: 12.0 Hz,lH),55.75-5.78(d,J: 11.2 Ηζ,ΙΗ) δ 5.14-5.17 (m,lH),5 1.27-1.35(m,6H). LCMS of Ci6Hi3F6N302(M+l)+:393.28 found 393.77 at 4.707 min (LCMS 99.25%).
[00720] General method for Example 76, Example 77, Example 78, Example 79, Example 83: A mixture of 5-(3-Chlorophenyl)-l,2,4-triazole (0.50 g, 3.4 mmol), respective propiolate (0.52 ml, 5.1 mmol) and some drops of triethylamine in acetonitrile under nitrogen was stirred at room temperature for 12-16 h. Acetonitrile was removed under reduced pressure to give a residual oil, which was purified by flash chromatography (3-5%> EtOAc/hexanes) to afford the both cis and trans isomers. Cis isomer was isolated 10-30%) and trans was isolated in 30-50%) with overall yield of 50-80%.
| WO2011109799A1 * |
Mar 5, 2011 |
Sep 9, 2011 |
Karyopharm Therapeutics, Inc. |
Nuclear transport modulatiors and uses thereof |
| US20110275607 |
Mar 5, 2011 |
Nov 10, 2011 |
Karyopharm Therapeutics, Inc. |
Nuclear transport modulators and uses thereof |
































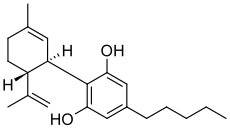 Cannabidiol
Seven Expanded Access INDs granted by FDA to U.S.
physicians to treat with Epidiolex 125 children suffering
from intractable epilepsy syndromes -
Cannabidiol
Seven Expanded Access INDs granted by FDA to U.S.
physicians to treat with Epidiolex 125 children suffering
from intractable epilepsy syndromes -