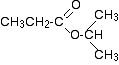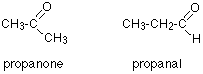Tout sur les médicaments הכל על תרופות كل شيئ عن الأدوية Все о наркотиках 关于药品的一切 డ్రగ్స్ గురించి అన్ని 마약에 관한 모든 것 Όλα για τα Ναρκωτικά Complete Tracking of Drugs Across the World by Dr Anthony Melvin Crasto, Worldpeacepeaker, worlddrugtracker, PH.D (ICT), MUMBAI, INDIA, Worlddrugtracker, Helping millions, 9 million hits on google on all websites, 2.5 lakh connections on all networks, “ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
C5H10O
Rule 2, omit O, gives C5H10
5 – 10/2 + 1 = 1 degree of unsaturation.
Look for 1 pi bond or aliphatic ring.
The band at 1727 indicates a carbonyl, probably an aldehyde; an aldehyde is also suggested by the band at 2719 which is likely the C-H stretch of the H-C=O group. The bands at 3000-2850 indicate C-H alkane stretches.
n
Proton NMR Spectrum
Since the IR spectrum indicates an aldehyde, look for this functionality in the NMR spectrum. The aldehydic proton appears in the NMR from 9-10, usually as a small singlet.
The spectrum above shows a small singlet corresponding to one proton at 9.2 ppm, confirming that the compound is an aldehyde. Protons on the carbon adjacent to the aldehyde carbonyl will show up at 2-2.7 ppm; this is the triplet peak of 2 protons at 2.4 ppm on the above spectrum. Thus, so far we know that there is an aldehyde group next to a methylene group which is next to a carbon that has two hydrogens:
This accounts for 3 of the 5 carbons in the molecule. The un-colored hydrogens in the above structure could correspond to the peak of 2 hydrogens centered at 1.6 ppm; this peak is a pentet indicating that these protons are adjacent to carbons with a total of 4 hydrogens. The peak centered at 1.35 ppm has two hydrogens and is a sextet, indicating it is next to carbons that have a total of 5 hydrogens. Finally, the peak at 0.9 ppm has 3 hydrogens and is a triplet, indicating it is a methyl group adjacent to a carbon that has 2 hydrogens. Therefore, it looks like the molecule is a straight-chain of 5 carbons with the aldehyde group at one end:
Note that the closer a group is to the carbonyl function, the further downfield it is shifted. Here is how the NMR correlates to the structure:
Structure of simian virus very similar to human. / SUPERSTOCK / AGE FOTOSTOCK
Three days is what it takes for the virus simian immunodeficiency (SIV), the most similar to HIV, to reach reservoirs (cells in which it is to siemrpe) microorganism. The measurement, which made scientific accuracy of Beth Israel Deaconess Medical Center in Boston, is key to preventing the affected macaque becomes a carrier for life VIS. The study sheds light on what happens in humans with HIV, and explains some of the latest achievements and disappointments that research has this infectious agent in recent years. The work is published in the latest edition ofNature.
– the reduction of carbonyl compounds is one of the most important synthetic reactions
– the catalytic enantioselective reduction of C=O has been achieved using:
* chiral oxazaborolidines and other related boronates (H3B as a source of hydrogen)
* transition metal catalysts (H2 as a source of hydrogen)
– first described by Itsuno et al. who observed that valinol reacts with 1 mol eq. of borane by producing 1 ml eq. of hydrogen gas and giving rise to the alkoxyborane derivative shown below:
– the aminoalkoxyborane derivatives (A and B) shown below are a result of the reaction of valinol with 2 eq. of borane (producing 2 mol eq. of hydrogen gas)
– the resulting aminoalkoxyborane (A or B) was found to catalyze the enantioselective reduction of PhCOMe
– the optical yield of the reduction was found to depend on the relative amounts of valinol and borane
– maximum optical yeild is reached with a borane-valinol ratio of 2.0
– the optical yeild remains almost constant within the borane-valinol ratio range of 2.0-3.0
– Itsuno et al. observed significantly higher optical yeilds when the hydrogens attached to the carbon atom of the terminal hydroxyl group were replaced by bulky groups, such as phenyls
– in the case of ketones other than aromatic ones optical yeilds were lower
– the optical yeild increases with the increasing difference in the volume of the substituents of the ketone
– an unusual relation between the optical yeild and reaction temperature was observed (studied by Itsuno et al. using methyl-tert-butyl ketone as a test system)
– the catalyst was found to work with more efficiency near 0 °C than at -78 °C
– reductions of functionalized ketones were studied
Other related reductions: (by Itsuno et al.)
– best optical yeilds were observed in the case of halohydrin formation
– the halohydrins were converted to form optically active epoxides without rasemization
– the reduction works best with chlorinated acetophenones
– two years latter Corey et al. developed the ideas of Itsuno et al. further and described a new and better catalyst (an oxazaborolidine derived from diphenylprolinol)
– the oxazaborolidine derived from diphenylprolinol gave better enantioselectivities for arylalkyl ketones than diphenylvalinol based derivatives
– Corey et al. proposed a mechanism for the catalytic reduction
– proline based oxazaborolidines are also known as CBS (Corey,Bakshi,Shibata) catalysts
– the better performance of CBS catalysts, relative to the performance of valinol-based catalysts, was related (by Corey et al.) to the higher angle strain on the partial B=N double bond at the 5,5-ring fusion
– the angle strain disturbs PI-resonance (A) and exposes the lone pair of the nitrogen atom (B) for borane to coordinate
– in THF (needed to stabilize highly polar reactive intermediates) the borane atom is not totally coordinated to the catalyst:
– the bicyclic (CBS) catalyst is capable of binding the borane more tightly than the related monocyclic system
– the more strained the B-N bond, the higher the proportion of catalyst present as a borane complex (ready to operate as a chiral catalyst)
– computational studies on the CBS catalyst indicate that not all atoms adjacent to the borane and nitrogen atoms of the partial B=N bond lie in the same plane (for the related torsion angles at 0 °C/180 °C +/- 22 °C see THA3,1563(1992))
– similar distortions were not observed with monocyclic oxazaborolidines
– the rigidity of the structure of CBS catalysts would also orient the borane to coordinate selectively on one of the faces of the oxazaborolidine ring
– coordination on the faces would involve:
* an attack on the less hindered side of the ring system (kinetic control)
* the formation of a 5,5,-cis-fused ring system is favoured over that of the highly strained 5,5-trans-fused system (thermodynamic control)
– in the formation of borane adducts of CBS catalysts only one adduct (lowering angle strain) is formed selectively
– other isomers of borane-oxazaborolidine adducts have also been considered, e.g.
– the system containing a hydride-bridged 6-ring was found to be more stable than the other diborane adducts
– the formation of hydride-bridged adducts indicates that the hydrogens of borons “scramble” in a mixture of borane and oxazaborolidine(s)
– this hydrogen – deuterium exchange “scrambling” has been observed experimentally [Tlahuext and Contreras, THA5, 395 (1994)]
– an X-ray study on a borane adduct of a CBS catalyst (a B-methylated derivative) proves that the borane atom coordinates to the nitrogen atom
– the X-ray structure of the N-adduct proves that the formation of N-adducts is possible and probably even favoured over the other adducts; nevertheless, the involvement of borane O-adducts of oxazaborolidines (as reactive intermediates) cannot completely be ruled out
– the mechanism of catalysis in the case of monocyclic systems has been proposed to be controlled by factors partially different from those controlling CBS catalysis
– the selectivity of the formation of borane cis/trans-adducts of monocyclic oxazaborolidines (e.g. those derived from valinols) has been calculated to be too low to fit the experimentally observed enantioselectivities, e.g. in the case of the simple model shown below:
– computational studies on simple models imply that the next step in the mechanistic cycle of catalysis should show significant selectivity
…………
– one of the most significant consequences of the N-coordination of borane to an oxazaborolidine is the substantially enhanced acidity of the ring boron [intramolecular stabilization through the partial PI-bond between the boron and adjacent nitrogen atom is not possible in the N-adduct]
– computational studies on the formation of N-, O- and N,O-(di)adducts (related to LUMO energies) imply that the parent oxazaborolidine is the weakest Lewis acid (highest LUMO energy), the borane N,O-diadduct being the strongest (lowest LUMO energy)
– the more the borane coordinates to the N- and O- atoms of an oxazaborolidine ring the less the ring boron is stabilized by partial PI-bonding
– not only are there differences in the Lewis acidities of the borane N- and O-adducts, but there are also many possible orientations from which a Lewis base (in this case a ketone) can best approach the ring boron
– in the case of borane N-adducts the orientation of the dipole moment favours the coordination of ketones
– in the case of borane O-adducts the orientation of the dipole moment is not particularly favourable; the incoming Lewis base has to approach the ring boron in the plane of the ring (this inhibits binding)!
– the orientation of the dipole moment of the borane N-adduct of the parent oxazaborolidine implies that the ketone (or any Lewis base) could react to form a borane-ketone cis-adduct
– the 5,5-diphenyl substituents direct the ketone to favour the anti-conformation over the syn-conformation (see the figure below)
– the structures of both the syn- and anti-adducts a borane-formaldehyde complex coordinating to the parent oxazaborolidine have been generated and assessed using computational methods
– these simple models, extended with two phenyl groups on C-5 of the oxazaborolidine ring (the orientations of the phenyls were set on the basis of the orientations of the corresponding hydrogens), show how hindered the syn-conformation is in the case of oxazaborolidines bearing bulky substituents on C-5
– plausible conformations of both the anti- and syn-adducts and the related transition states of the hydride transfer have been studied computationally
– the hydride transfer taking place in the borane-ketone adducts of oxazaborolidines has been proposed to lead to the formation of an intramolecular adduct of an alkoxyborane, which in turn results in the formation of an aminoborane (A)
– the aminoborane can react further to form an oxazadiboretane (structure B)
– the oxazaboretane system (B) may undergo a number of reactions, one of which leads to the regeneration of the catalyst, whereas another leads to the formation of an alkoxyborane adduct analogous to the original borane adduct of oxazaborolidine
– NMR studies performed on the products of the related stoichiometric reduction carried out in the presence of Et3N gave the alkoxyborane
– a few other examples:
– further NMR studies on the products formed in the reduction of acetaldehyde with the same catalyst led to the structural interpretations shown below:
– on the basis of data obtained with 13C-NMR studies, it is not clear whether the species the signals originate from are oxazadiboretanes or their related openchain isomers (of which the latter ones are shown in the figure above)
– the results indicate that the first (of the two) hydride transfer(s) occurs with higher enantioselectivity than the second
– in addition to the mechanism of the regeneration of the catalyst discussed above, another plausible pathway has been proposed on the basis of computational studies carried out on hydride-bridged adducts of borane coordinated to oxazadiboretanes
– the regeneration of oxazaborolidine catalysts used in the enantioselective reduction of ketones was proposed to involve the hydride-bridged adduct shown below (two conformers; H2C=O as a model of the ketone and the parent oxazaborolidine as a model of the catalyst)
– the energies of the insertion of borane into oxazadiboretanes are rather low relative to those involved with most energy requiring/liberating steps in the reduction
…………
– the latest mechanistic proposal (on the basis of a computational AM1 study) is shown below:
– in contrast to the results of NMR studies on the model reaction of CBS reduction (H3C-CHO as a model of ketones), an alkoxyborane adduct (structurally analogous to that of the related borane adduct) is not included in the mechanism
– although the mechanism of the CBS reduction is not completely clear at this time, the stereochemical outcome of the reduction can easily be predicted
– boranes other than BH3 can also be used as a source of hydrogen in CBS reductions; e.g. catecholborane shown below
– in addition to the enantioselective synthesis of epoxides (Itsuno et al.), the products of these enantioselective reductions have been converted to many valuable compounds
a) The enantioselective synthesis of ALPHA-amino-acids (including unnatural ones)
b) The enantioselective synthesis of ALPHA-hydroxy-acids
c) The enantioselective synthesis of 1-deuterio primary alcohols
d) The enantioselective synthesis of benzylic thiols
e) The enantioselective synthesis of oxiranes
– in the case of CBS reductions, the coordination site of the ketone is usually determined by the difference in bulkiness of the substituents (RL and RS), but other selection mechanisms also exist
– any effect making one of the two lone pairs on the carbonyl oxygen atom of a ketone more basic than the other should work, e.g. CBS reduction of benzophenones
– the lone pair “a” (trans to the donating group) should be more basic
– two transition states:
– the formation of A (stabililzed by PI-electron donation from the p-OR group) should be favoured over B
– the stereochemical outcome of the reduction corresponds to the transition state A
– high selectivities are observed (although both substituents of the ketone being reduced are almost equally bulky)
– polymer-bound chiral oxazaborolidines have also been shown to work with enantioselectivities similar to those of their free monomeric analogs, e.g.
– the reduction of acetophenone using this polymeric catalyst gave 95% ee (the corresponding monomeric catalyst gave 97% ee)
– it has been shown that oxazaborolidines of which the basicity of the ring nitrogen has been reduced can also be utilized in the enantioselective reduction performed using oxazaborolidines
– in these catalysts:
* the basicity of the ring nitrogen has been reduced by an electron withdrawing substituent (e.g. Me-SO2)
* one of the bulky 5,5-substituents has been removed (one face of the ring has been made more accessible than the other)
* the bulky 5-substituent of the H3B O-adduct affords an axial conformation in which it:
a) is almost orientated against the plane of the sp2-hybridized oxygen of the ring, e.g.
b) substituents 4 and 5 are trans about the ring (otherwise repulsive interactions between the substituents will exist)
the substituents of the ring:
c) will block one face of the borane-oxazaborolidine O-adduct (the incoming ketone can approach the Lewis acidic boron with greater ease than from the face opposite the 5-substituent)
d) will direct the coordination of the incoming ketone towards an equatorial conforamation [the substituent of the boron (e.g. H) being cis to the bulky 5-substituent]
e) orient the coordinated H3B optimally (if the 5-substituent is in an equatorial conformation, the borane bound to the ring oxygen will reside far from the carbonyl carbon of the coordinated ketone)
(a complex in which the ketone is in an equatorial conformation would have even more problems)
UncategorizedComments Off on Identification and synthesis of a male-produced pheromone for the neotropical root weevil diaperpes abbreviatus (coleoptera: curculionidae) US 20130189222 A1
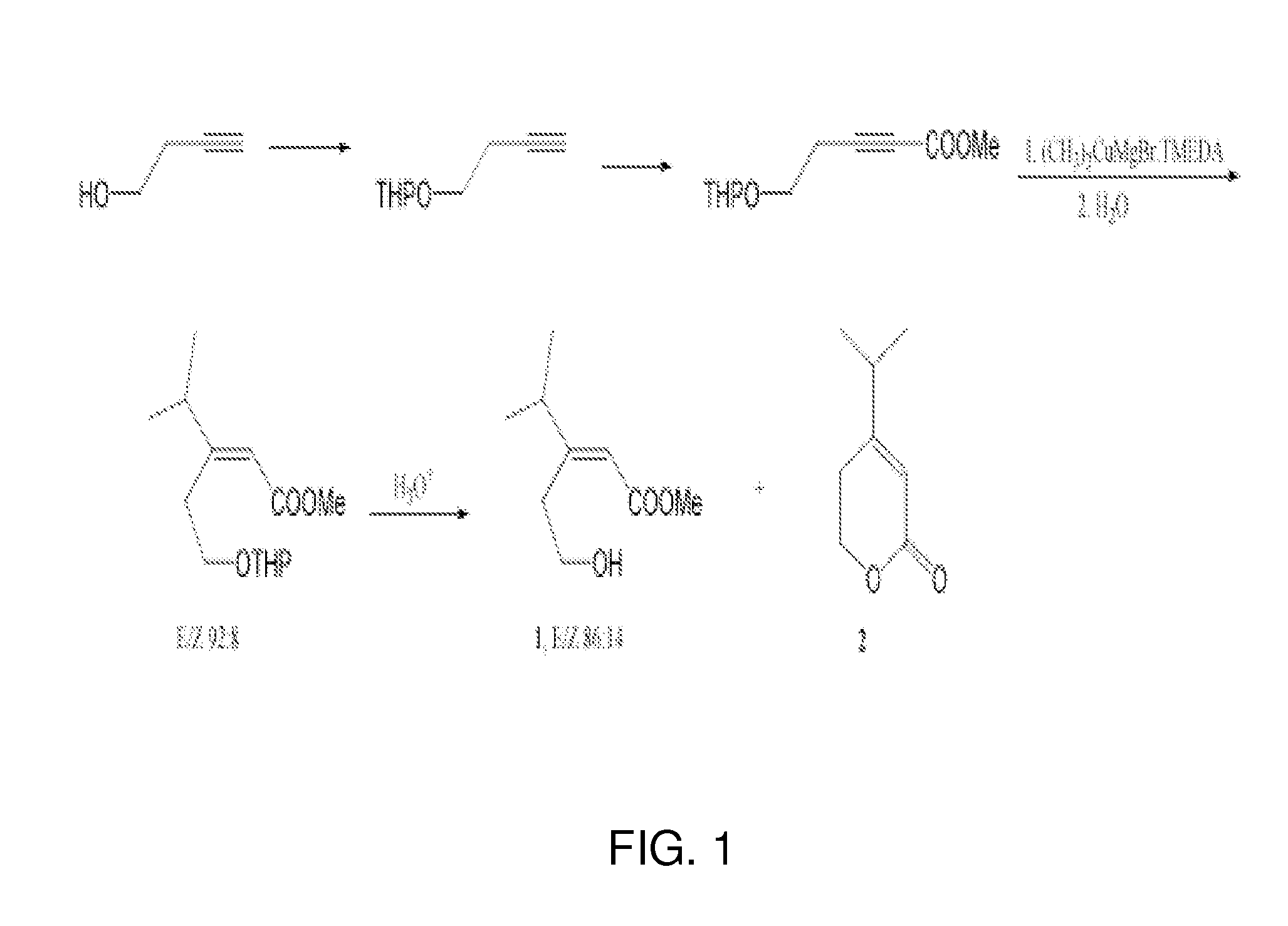
an unsaturated hydroxy ester pheromone collected from the headspace and feces of male Diaprepes abbreviatus was isolated, identified and synthesized. The pheromone, methyl (E)-3-(2-hydroxyethyl)-4-methyl-2-pentenoate, was discovered by gas chromatography-coupled electroantennogram detection (GC-EAD) and identified by gas chromatography-mass spectrometry (GC-MS) and nuclear magnetic resonance spectroscopy (NMR). The synthetic protocol yielded a 86:14 mixture of methyl (E)-3-(2-hydroxyethyl)-4-methyl-2-pentenoate and an inactive methyl (Z)-3-(2-hydroxyethyl)-4-methyl-2-pentenoate along with a lactone decomposition product. The activity of the synthetic E isomer was confirmed by GC-EAD, GC-MS, NMR and behavioral assays. No antennal response was observed to the Z isomer or the lactone. In a two-choice olfactometer bioassay, female D. abbreviatus moved upwind towards the synthetic pheromone or a source of natural pheromone more often as compared to clean air. Males showed no clear preference for the synthetic pheromone.
The root weevil Diaprepes abbreviatus (L.), is a major pest of citrus in the Caribbean and Florida. Prior to the 1960’s, D. abbreviatus was reported only in the Caribbean. Because multiple phenotypic populations occur on Puerto Rico it is suggested that D. abbreviatus originated in Puerto Rico (Lapointe 2004). Since its discovery near Apopka, Fla. in 1964, it has spread to Louisiana, Texas and California. There is no geographic or climatic barrier to prevent the southern movement of this insect to Mexico, Mesoamerica and South America (Lapointe et al. 2007).
This migration is of concern because this insect is destructive. Adult beetles of D. abbreviatus oviposit and feed on leaves of a wide range of hosts including more than 270 species of plants in 59 plant families. Feeding by adults on leaves causes a characteristic notching pattern; however, the larval stage causes the most serious damage. Neonate larvae fall to the ground and burrow into the soil where they feed on progressively larger roots over a period of months as they grow. Larval feeding on citrus tree roots can eventually girdle the crown area of the root system, killing the host plant. When larval development is completed, adults emerge from the soil to feed upon foliage where aggregation, mating and oviposition take place. In certain citrus growing areas, root damage by larval D. abbreviatus creates favorable conditions for species ofPhytophthora, a very serious and often lethal plant pathogen, to invade roots and further hasten the decline of trees.
In Florida, citrus growers spend up to $400/acre for combined control of D. abbreviatus and Phytophthora. In 2009, it was estimated that the total increase in costs per ton due to the establishment and spread of Diaprepes root weevil in California would be $53.60 for orange, $45.20 for grapefruit, $42.50 for lemon and $200.00 for avocado. In view of the negative economic impact caused by the feeding of this insect and in view of the fact that there appear to be no natural barriers to important agricultural citrus growing areas, attractants that will allow for the monitoring, tracking, trapping and destroying of this insect have been sought.
Diaprepes abbreviatus has been placed in the subfamily Entiminae of the Curculionidae (Marvaldi et al. 2002) Within the superfamily Cu rculionoidea (weevils) the majority of attractants or pheromones identified to date are long-range, male-produced aggregation pheromones (Seybold and Vanderwel 2003, Ambrogi et al. 2009). Aggregation of D. abbreviatus adults and the occurrence of so-called “party trees” have been observed (Wolcott 1936). Schroeder (1981) suggested a male-produced pheromone attracted females and a female-produced pheromone attracted males. Beavers et al. (1982) showed in laboratory tests that male and female D. abbreviatus were significantly attracted to the frass of the opposite sex. Jones and Schroeder (1984) demonstrated a male-produced pheromone in the feces that attracted both sexes. A pheromone responsible for arrestment behavior was suggested by Lapointe and Hall (2009). U.S. Pat. No. 8,066,979 to Dickens et al. showed for the first time that D. abbreviatus adults have olfactory receptors for secondary plant metabolites that belong to diverse chemical groups: (a) alcohol and aldehyde monoterpenes (e.g., linalool, citronellal, nerol, and trans-geraniol), (b) green leaf volatiles (e.g., cis-3-hexen-1-ol and trans-2-hexen-1-ol), and (c) an aromatic monoterpenoid (e.g., carvacrol). Otálora-Luna et al. (2009) identified by gas-chromatograph electroantennograph detection (GC-EAD) a number of plant volatiles from citrus leaves that elicited antennal response in D. abbreviatus. Such kairomones may act in concert with a pheromone to attract conspecifics to a suitable food source (Dickens 1990). Only one pheromone, that of Sitona lineatus (4-methyl-3,5-heptanedione), an aggregation pheromone, has been isolated from the Entiminae (broad-nosed weevils) (Blight et al. 1984). Blight and Wadhams (1987) suggested that S. lineatus produces its aggregation pheromone in the spring and that the pheromone activity is synergized by host plant volatiles including (Z)-3-hexen-1-ol and linalool.
chromatographed again with hexanes/ethyl acetate/MeOH, 16:6:1 to furnish 1 (E/Z 86:14, approximately 90 mg, approximately 58%) in the less polar fraction.
Developed by Vertex Pharmaceuticals, it is the first drug that attacks not just the symptoms but the underlying cause of cystic fibrosis, a genetic lung disease that usually kills victims by the time they reach their 40s. It doesn’t work for every sufferer of the disease, but rather for a small subset — probably around 2,000 people — who have a specific genetic mutation that the drug targets. But for those it helps, it is life changing. text clipped read at
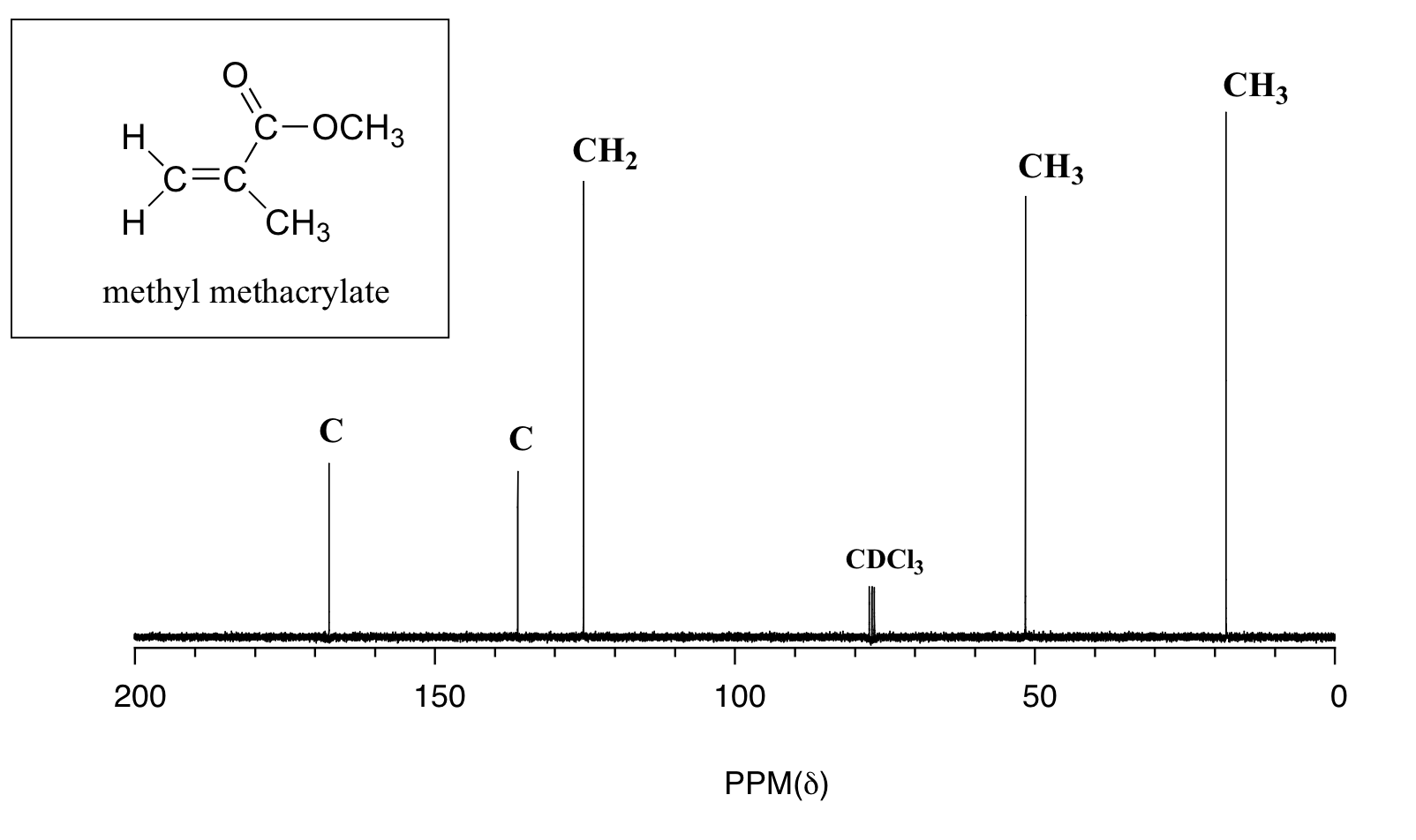
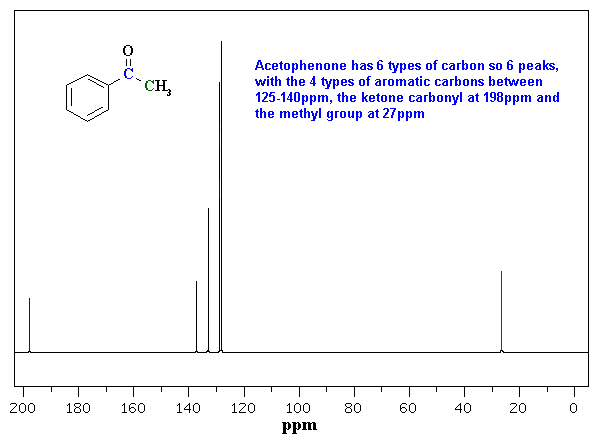
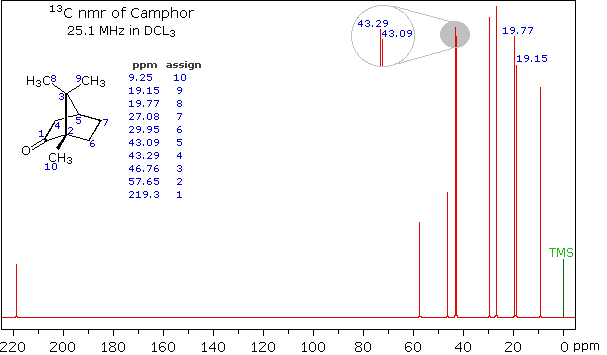
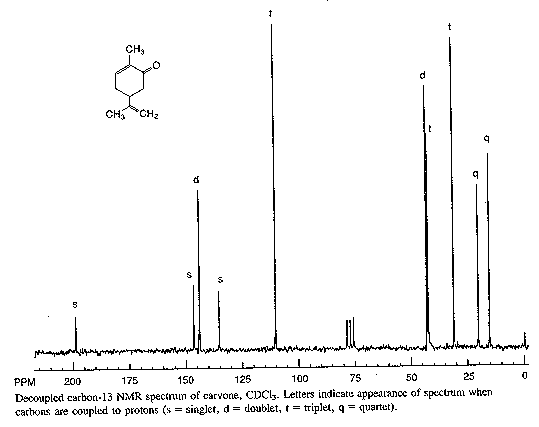
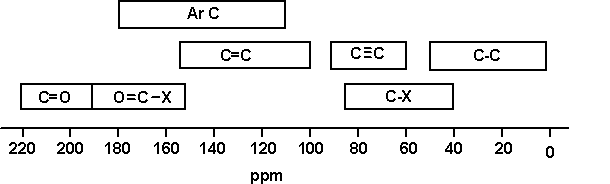
Here are a couple of examples for judging 13C chemical shifts:
Minimum description used……..mostly pictorial
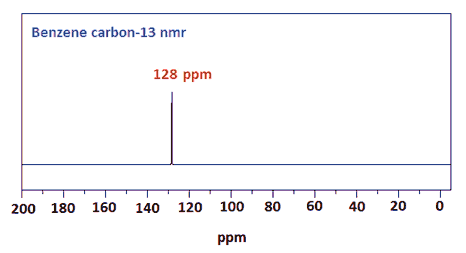
Start with benzene
Then toluene
Remember that 13C shifts generally follow the pattern of 1H shifts, but are much larger.
Thus, in (a)
The peak at lowest field is the caarbon with the OH attached
The peak at highest field is the CH3 farthest from the OH
Generally in both 13C and 1H spectra, CH2 groups fall to lower field than CH3, which assigns the remaining peaks.
For (b):
The peak at lowest field is the C=O carbon, shifted by electronegativity and “diamagnetic anisotropy”
Assignment of the other two peaks follows from the CH2 lower than CH3 rule.
Now try predicting the splitting pattern to be found if the decoupler is turned off.
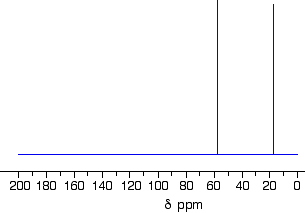
Taking a close look at three C-13 NMR spectraThe C-13 NMR spectrum for ethanol
Remember that each peak identifies a carbon atom in a different environment within the molecule. In this case there are two peaks because there are two different environments for the carbons.The carbon in the CH3 group is attached to 3 hydrogens and a carbon. The carbon in the CH2 group is attached to 2 hydrogens, a carbon and an oxygen.So which peak is which?You might remember from the introductory page that the external magnetic field experienced by the carbon nuclei is affected by the electronegativity of the atoms attached to them. The effect of this is that the chemical shift of the carbon increases if you attach an atom like oxygen to it. That means that the peak at about 60 (the larger chemical shift) is due to the CH2 group because it has a more electronegative atom attached.
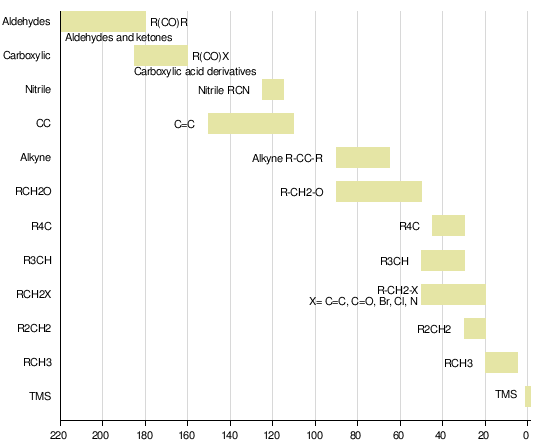
A table of typical chemical shifts in C-13 NMR spectra
carbon environment
chemical shift (ppm)
C=O (in ketones)
205 – 220
C=O (in aldehydes)
190 – 200
C=O (in acids and esters)
170 – 185
C in aromatic rings
125 – 150
C=C (in alkenes)
115 – 140
RCH2OH
50 – 65
RCH2Cl
40 – 45
RCH2NH2
37 – 45
R3CH
25 – 35
CH3CO-
20 – 30
R2CH2
16 – 25
RCH3
10 – 15
In the table, the “R” groups won’t necessarily be simple alkyl groups. In each case there will be a carbon atom attached to the one shown in red, but there may well be other things substituted into the “R” group.
If a substituent is very close to the carbon in question, and very electronegative, that might affect the values given in the table slightly.
For example, ethanol has a peak at about 60 because of theCH2OH group. No problem!
It also has a peak due to the RCH3 group. The “R” group this time is CH2OH. The electron pulling effect of the oxygen atom increases the chemical shift slightly from the one shown in the table to a value of about 18.
A simplification of the table
At the time of writing, a draft UK syllabus (Cambridge pre-U) was expecting their students to learn the following simplification:
carbon environment
chemical shift (ppm)
C-C
0 – 50
C-O
50 – 100
C=C
100 – 150
C=O
150 – 200
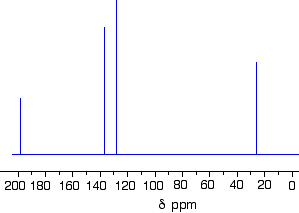
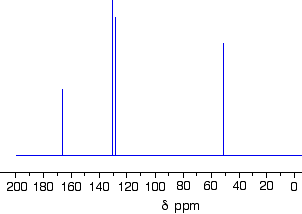
The C-13 NMR spectrum for but-3-en-2-oneThis is also known as 3-buten-2-one (amongst many other things!)
Here is the structure for the compound:
You can pick out all the peaks in this compound using the simplified table above.
The peak at just under 200 is due to a carbon-oxygen double bond. The two peaks at 137 and 129 are due to the carbons at either end of the carbon-carbon double bond. And the peak at 26 is the methyl group which, of course, is joined to the rest of the molecule by a carbon-carbon single bond.
If you want to use the more accurate table, you have to put a bit more thought into it – and, in particular, worry about the values which don’t always exactly match those in the table!
The carbon-oxygen double bond in the peak for the ketone group has a slightly lower value than the table suggests for a ketone. There is an interaction between the carbon-oxygen and carbon-carbon double bonds in the molecule which affects the value slightly. This isn’t something which we need to look at in detail for the purposes of this topic.
You must be prepared to find small discrepancies of this sort in more complicated molecules – but don’t worry about this for exam purposes at this level. Your examiners should give you shift values which exactly match the compound you are given.
The two peaks for the carbons in the carbon-carbon double bond are exactly where they would be expected to be. Notice that they aren’t in exactly the same environment, and so don’t have the same shift values. The one closer to the carbon-oxygen double bond has the larger value.
And the methyl group on the end has exactly the sort of value you would expect for one attached to C=O. The table gives a range of 20 – 30, and that’s where it is.
One final important thing to notice. There are four carbons in the molecule and four peaks because they are all in different environments. But they aren’t all the same height. In C-13 NMR, you can’t draw any simple conclusions from the heights of the various peaks.
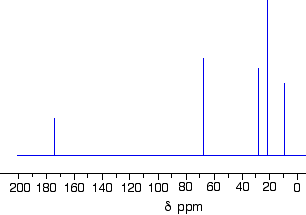
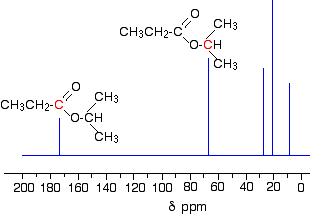
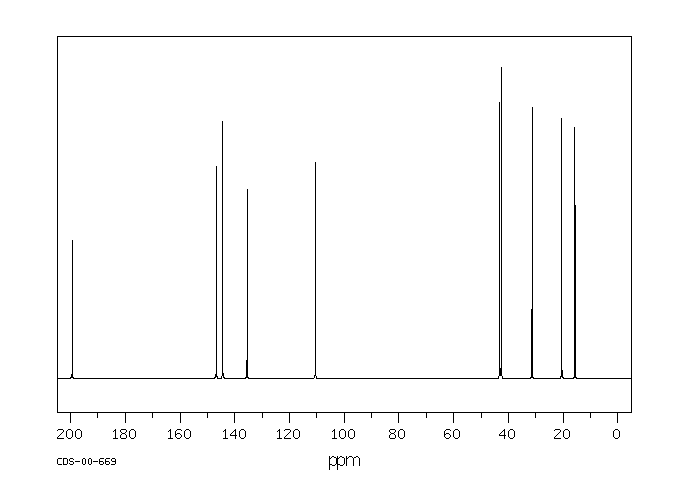
The C-13 NMR spectrum for 1-methylethyl propanoate
1-methylethyl propanoate is also known as isopropyl propanoate or isopropyl propionate.
Here is the structure for 1-methylethyl propanoate:
Two simple peaks
There are two very simple peaks in the spectrum which could be identified easily from the second table above.
The peak at 174 is due to a carbon in a carbon-oxygen double bond. (Looking at the more detailed table, this peak is due to the carbon in a carbon-oxygen double bond in an acid or ester.)
The peak at 67 is due to a different carbon singly bonded to an oxygen. Those two peaks are therefore due to:
If you look back at the more detailed table of chemical shifts, you will find that a carbon singly bonded to an oxygen has a range of 50 – 65. 67 is, of course, a little bit higher than that.
As before, you must expect these small differences. No table can account for all the fine differences in environment of a carbon in a molecule. Different tables will quote slightly different ranges. At this level, you can just ignore that problem!
Before we go on to look at the other peaks, notice the heights of these two peaks we’ve been talking about. They are both due to a single carbon atom in the molecule, and yet they have different heights. Again, you can’t read any reliable information directly from peak heights in these spectra.
The three right-hand peaks
From the simplified table, all you can say is that these are due to carbons attached to other carbon atoms by single bonds. But because there are three peaks, the carbons must be in three different environments.
The more detailed table is more helpful.
Here are the structure and the spectrum again:
The easiest peak to sort out is the one at 28. If you look back at the table, that could well be a carbon attached to a carbon-oxygen double bond. The table quotes the group as CH3CO-, but replacing one of the hydrogens by a simple CH3 group won’t make much difference to the shift value.
The right-hand peak is also fairly easy. This is the left-hand methyl group in the molecule. It is attached to an admittedly complicated R group (the rest of the molecule). It is the bottom value given in the detailed table.
The tall peak at 22 must be due to the two methyl groups at the right-hand end of the molecule – because that’s all that’s left. These combine to give a single peak because they are both in exactlythe same environment.
If you are looking at the detailed table, you need to think very carefully which of the environments you should be looking at. Without thinking, it is tempting to go for the R2CH2 with peaks in the 16 – 25 region. But you would be wrong!
The carbons we are interested in are the ones in the methyl group, not in the R groups. These carbons are again in the environment: RCH3. The R is the rest of the molecule.
The table says that these should have peaks in the range 10 – 15, but our peak is a bit higher. This is because of the presence of the nearby oxygen atom. Its electronegativity is pulling electrons away from the methyl groups – and, as we’ve seen above, this tends to increase the chemical shift slightly.
Once again, don’t worry about the discrepancies. In an exam, perhaps your examiners will just want you to have learnt the simple table above – in which case, they can’t expect you to work out which peak is which in a complicated spectrum of this sort. Or they will give you tables of chemical shifts – in which case, they will give you values which match the peaks in the spectra.
Remember that you are only doing an introduction to C-13 NMR at this level. It isn’t going to be that hard in an exam!
Working out structures from C-13 NMR spectra
So far, we’ve just been trying to see the relationship between carbons in particular environments in a molecule and the spectrum produced. We’ve had all the information necessary. Now let’s make it a little more difficult – but we’ll work from much easier examples!
In each example, try to work it out for yourself before you read the explanation.
Example 1
How could you tell from just a quick look at a C-13 NMR spectrum (and without worrying about chemical shifts) whether you had propanone or propanal (assuming those were the only options)?
Because these are isomers, each has the same number of carbon atoms, but there is a difference between the environments of the carbons which will make a big impact on the spectra.
In propanone, the two carbons in the methyl groups are in exactly the same environment, and so will produce only a single peak. That means that the propanone spectrum will have only 2 peaks – one for the methyl groups and one for the carbon in the C=O group.
However, in propanal, all the carbons are in completely different environments, and the spectrum will have three peaks.
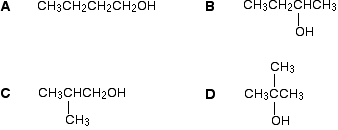
Example 2
Thare are four alcohols with the molecular formula C4H10O.
Which one produced the C-13 NMR spectrum below?
You can do this perfectly well without referring to chemical shift tables at all.
In the spectrum there are a total of three peaks – that means that there are only three different environments for the carbons, despite there being four carbon atoms.
In A and B, there are four totally different environments. Both of these would produce four peaks.
In D, there are only two different environments – all the methyl groups are exactly equivalent. D would only produce two peaks.
That leaves C. Two of the methyl groups are in exactly the same environment – attached to the rest of the molecule in exactly the same way. They would only produce one peak. With the other two carbon atoms, that would make a total of three. The alcohol is C.
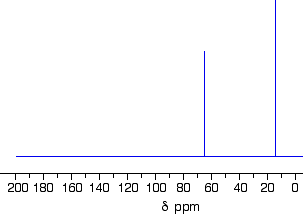
Example 3
This follows on from Example 2, and also involves an isomer of C4H10O but which isn’t an alcohol. Its C-13 NMR spectrum is below. Work out what its structure is.
Because we don’t know what sort of structure we are looking at, this time it would be a good idea to look at the shift values. The approximations are perfectly good, and we will work from this table:
carbon environment
chemical shift (ppm)
C-C
0 – 50
C-O
50 – 100
C=C
100 – 150
C=O
150 – 200
There is a peak for carbon(s) in a carbon-oxygen single bond and one for carbon(s) in a carbon-carbon single bond. That would be consistent with C-C-O in the structure.
It isn’t an alcohol (you are told that in the question), and so there must be another carbon on the right-hand side of the oxygen in the structure in the last paragraph.
The molecular formula is C4H10O, and there are only two peaks. The only solution to that is to have two identical ethyl groups either side of the oxygen.
The compound is ethoxyethane (diethyl ether), CH3CH2OCH2CH3.
Example 4
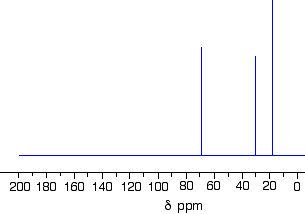
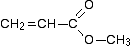
Using the simplified table of chemical shifts above, work out the structure of the compound with the following C-13 NMR spectrum. Its molecular formula is C4H6O2.
Let’s sort out what we’ve got.
There are four peaks and four carbons. No two carbons are in exactly the same environment.
The peak at just over 50 must be a carbon attached to an oxygen by a single bond.
The two peaks around 130 must be the two carbons at either end of a carbon-carbon double bond.
The peak at just less than 170 is the carbon in a carbon-oxygen double bond.
Putting this together is a matter of playing around with the structures until you have come up with something reasonable. But you can’t be sure that you have got the right structure using this simplified table.
In this particular case, the spectrum was for the compound:
If you refer back to the more accurate table of chemical shifts towards the top of the page, you will get some better confirmation of this. The relatively low value of the carbon-oxygen double bond peak suggests an ester or acid rather than an aldehyde or ketone.
It can’t be an acid because there has to be a carbon attached to an oxygen by a single bond somewhere – apart from the one in the -COOH group. We’ve already accounted for that carbon atom from the peak at about 170. If it was an acid, you would already have used up both oxygens in the structure in the -COOH group.
Without this information, though, you could probably come up with reasonable alternative structures. If you were working from the simplified table in an exam, your examiners would have to allow any valid alternatives.
Figure 3: 1H and 13C NMR spectra of the abcc-monomer and the ABCC-sequence-regulated copolymer.
(a) 1H and 13C NMR spectra of 7. (b) 1H and 13C NMR spectra of poly(7) obtained with CuCl/N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA) ([CuCl]0=200 mM; [PMDETA]0=800 mM) in bulk at 100 °C. All spectra were measured in CDCl3 at room temperature The brackets for repeating units in poly(7) are positioned differently from those in Figs. 1 and 4 so that the signals originating from the terminal groups can be assigned to the chemical structures.
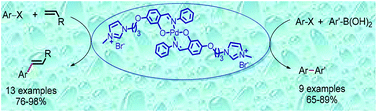
polymorphComments Off on DRUG SYNTHESIS USING SUZUKI RXN..Imidazolium ionic liquid-tagged palladium complex: an efficient catalyst for the Heck and Suzuki reactions in aqueous media
A highly recyclable ionic liquid-tagged palladium complex has been synthesized and used as an efficient catalyst for Suzuki and Heck couplings in aqueous media.
An air stable, water soluble, and efficient ionic liquid-tagged Schiff base palladium complex was prepared. The synthesized complex was well characterized by NMR, mass spectrometry, FT-IR, UV-visible spectroscopy and powder X-ray diffraction. The complex was used as a catalyst for the Suzuki and Heck cross-coupling reactions in water. Good to excellent yields were achieved using a modest amount of the catalyst. In addition, the catalyst can be easily reused and recycled for six steps without much loss in activity, exhibiting an example of sustainable and green methodology.
aPharmaceutical Analytical & Solid-State Chemistry Research Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
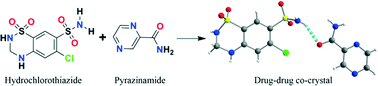
The drug-drug co-crystal of hydrochlorothiazide with pyrazinamide is a potential candidate for development of hydrochlorothiazide formulations for combinational therapy.
Crystal engineering principles were employed in designing new co-crystals of hydrochlorothiazide (HCT). A variety of potential co-crystal formers were initially identified in a search of the Cambridge Structural Database with complementary hydrogen-bonding functionalities. Subsequent co-crystallization screening monitored by powder X-ray diffraction led to the discovery of new crystalline phases of HCT with pyrazinamide (1), 4,4′-bipyridine (2), 1,2-bis(4-pyridyl)ethane (3), and trans-1,2-bis(4-pyridyl)ethylene (4). All of the resulting co-crystals were thoroughly characterized by X-ray diffraction, FT-IR and Raman spectroscopy, and thermal analysis. Noticeably, the co-crystal 1 involves the formation of drug–drug co-crystals of HCT and pyrazinamide, which makes it a potential candidate for development of HCT formulations for combinational therapy.
All about Drugs, live, by DR ANTHONY MELVIN CRASTO, Worlddrugtracker, Helping millions, 7 million hits on google, pushing boundaries, one lakh plus connections worldwide, 3 lakh plus VIEWS on this blog in 193 countries
















 n
n