











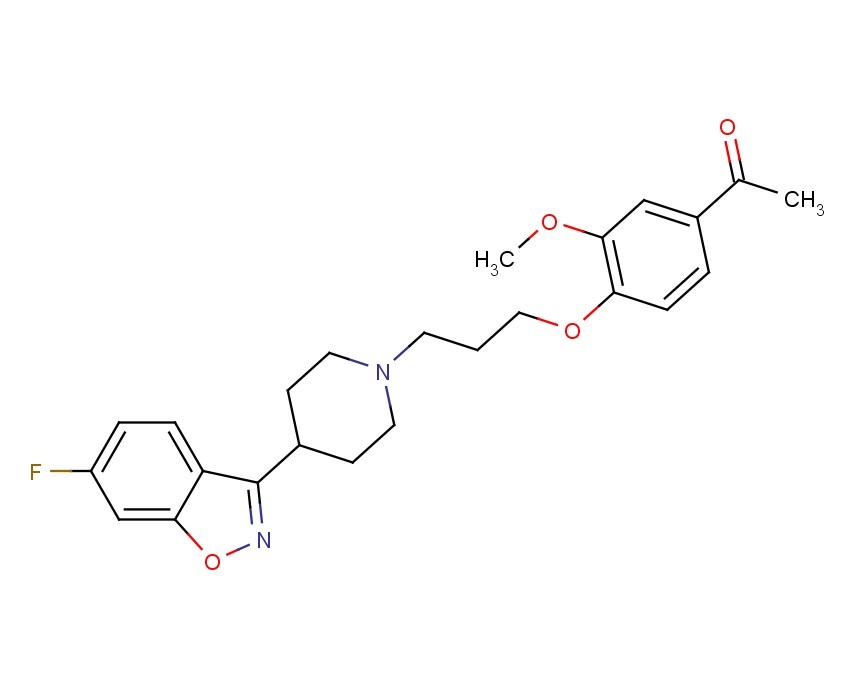
Iloperidone (Fanapt), ILO-522, HP-873, Zomaril, 133454-47-4, antipsychotic
1-[4-[3-[4-(6-Fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl]propoxy]-3-methoxyphenyl]ethanone; 1-[3-(4-Acetyl-2-methoxyphenoxy)propyl]-4-(6-fluoro-1,2-benzisoxazol-3-yl)piperidine; 4′-[3-[4-(6-Fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl]propoxy]-3′-methoxyacetophenone
Aventis Pharma (Originator), Novartis (Licensee), Titan (Licensee) Vanda Pharmaceuticals (Licensee)
Vanda Pharmaceuticals (Licensee)
Iloperidone(Fanapt) is a monoamine directed towards acting upon and antagonizing specific neurotransmitters, particularly multiple dopamine and serotonin receptor subtypes.
Schizophrenia is a chronic, severe, and debilitating mental disorder that affects approximately 2.4 million Americans, around 1.1% of the population. The net cost of this disorder is staggering as estimates from 2002 reveal this disorder to cost $62.7 billion. A major issue with the treatment of schizophrenia is that patients show varying levels of response and tolerance to available therapies. Although the symptoms of the disease are very severe, estimates show that approximately 3 out of 4 patients discontinue medication prior to completing 18 months of treatment, many times due to the severe side effects of the approved medications.
Synthesis
J.T. Strupczewski, K.J. Bordeau, Y. Chiang, E.J. Glamkowski, P.G.
Conway, R. Corbett, H.B. Hartman, M.R. Szewczak, C.A. Wilmot andG.C. Helsley, J. Med. Chem., 38, 1119 (1995).
US 4355037
V. Miklos, WO Patent 031497 (2010).
J.T. Strupczewski, EP Patent 0402644 (1990)
The product is protected by the U.S. Pat. No. 5,364,866, U.S. Pat. No. RE 39198 E and EP 402644 B1.U.S. Pat. No. 5,364,866 and U.S. Pat. No. 5,663,449.EP 542136, EP 612318, EP 730452, JP 95501055, JP 97511215, US 5364866, US 5776963, WO 9309102, WO 9511680.US 4355037,EP 0542136; EP 0612318; EP 0730452; EP 0957102; EP 0959075; EP 0959076; EP 0963984; JP 1995501055; JP 1997511215; US 5364866; US 5776963; WO 9309102; WO 9511680
The first reported synthetic method for Iloperidone is described in patent EP 402644 A1.
In U.S. Patent US5776963 and patent family EP4 (^ 644, there is disclosed a method for preparing iloperidone,
The synthetic method reported(4, 5) for 1 involves two chemical steps: O-alkylation of acetovanillone (2) with 1-bromo-3-chloropropane (3) to obtain chloro derivative 4 followed byN-alkylation of piperidine intermediate 5 with 4. The reported process for 4 comprises O-alkylation of 2 with 3 in acetone in the presence of potassium carbonate for 20 h to provide 4as an oil after usual work up, which was then vacuum (0.1 mmHg) distilled to collect desired product 4 at 141–143 °C with around 85% yield (Scheme 1, Path A). Some of the drawbacks of this process are as follows: longer reaction time (around 20 h), formation of 6–7% of dimer impurity (10, Scheme 2), high-vacuum distillation to achieve the quality, which is always a cumbersome process at industrial scale, requiring special apparatus and skill set, and degradation and charring of some portion of product during high-vacuum distillation. Further, the next step comprises N-alkylation of 4 with 5 in N,N-dimethylformamide (DMF) in the presence of potassium carbonate to provide iloperidone (1) as a crude solid, which was purified by crystallization using ethanol to yield pure 1 with 58% yield (Scheme 1, Path A). Some of the lacunae observed with the above process includes the following: (a) low yields, (b) formation of carbamate impurity 13 (Scheme 2) in the range 15–20% due to the use of potassium carbonate, (c) ineffective purification by crystallization using ethanol to eliminate carbamate impurity below 0.15%, and (d) iloperidone obtained by the above synthetic process was beige in color.



The synthetic route is as follows:
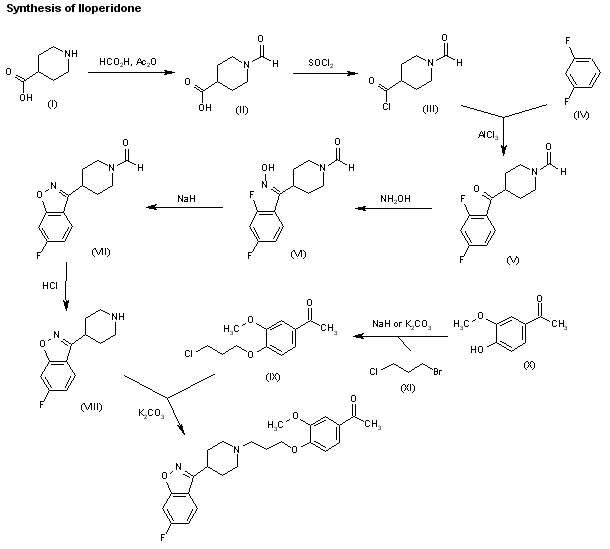
The reaction of piperidine-4-carboxylic acid (I) with formic acid and acetic anhydride gives 1-formylpiperidine-4-carboxylic acid (II), which is treated with SOCl2 and acetic anhydride to yield the corresponding acyl chloride (III). The Friedel-Crafts condensation of (III) with refluxing 1,3-difluorobenzene (IV) by means of AlCl3 affords 4-(2,4-difluorobenzoyl)-1-formylpiperidine (V), which is treated with hydroxylamine in refluxing ethanol to give the corresponding oxime (VI). The cyclization of (VI) by means of NaH in hot THF/DMF yields 6-fluoro-3-(1-formylpiperidin-4-yl)-1,2-benzisoxazole (VII), which is treated with HCl in refluxing ethanol to afford 6-fluoro-3-(4-piperidyl)-1,2-benzisoxazole (VIII). Finally, this compound is condensed with 4-(3-chloropropoxy)-3-methoxyacetophenone (IX) by means of K2CO3 in hot DMF. The intermediate 4-(3-chloropropoxy)-3-methoxyacetophenone (IX) can be obtained by condensation of 4-hydroxy-3-methoxyacetophenone (IX) with 3-chcloropropyl bromide (X) by means of NaH or K2CO3 in DMF.

Iloperidone, also known as Fanapt, Fanapta, and previously known as Zomaril, is an atypical antipsychotic for the treatment ofschizophrenia.
Accordingly, 6-fluoro-3-(4-piperidyl)-1,2-benzoxazole 1 and 1-[4-(3-chloropropoxy)-3-methoxy-phenyl]ethanone 2 were heated in presence of potassium carbonate using dimethylformamide solvent to afford 1-[4-[3-[4-(6-fluoro-1,2-benzoxazol-3-yl)-1-piperidyl]propoxy]-3-methoxy-phenyl]ethanone also called Iloperidone
It was approved by the U.S. Food and Drug Administration (FDA) for use in the United States on May 6, 2009.
It’s not yet approved in India.
Hoechst Marion Roussel Inc. made initial inquiries into the drug; however, in May 1996, they discontinued research, and in June 1997 gave research rights to Titan Pharmaceuticals. Titan then handed over worldwide development, manufacturing and marketing rights to Novartis in August 1998. On June 9, 2004, Titan Pharmaceuticals announced that the Phase III development rights have been acquired by Vanda Pharmaceuticals. The original launch date was scheduled for 2002. On November 27, 2007, Vanda Pharmaceuticals announced that the U.S. Food and Drug Administration (FDA) had accepted their New Drug Application for iloperidone, confirming the application is ready for FDA review and approval. On July 28, 2008, the FDA issued a “Not Approvable” letter to Vanda Pharmaceuticals concerning the drug, stating that further trials are required before a decision can be made concerning marketed usage of iloperidone.
Chemically designated as 1-[4-[3-[4-(6-fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl]propoxy]-3-methoxyphenyl]ethanone, is a second generation atypical antipsychotic agent. Iloperidone, also known as Fanapt, Fanapta, and Zomaril, was approved by the U.S. Food and Drug Administration (FDA) for use in the United States on May 6, 2009 and is indicated for the acute treatment of schizophrenia in adults. Iloperidone has been shown to act as an antagonist at all tested receptors. It was found to block the sites of noradrenalin (α2C), dopamine (D2A and D3), and serotonin (5-HT1A and 5-HT6) receptors.(2) In addition, pharmacogenomic studies identified single nucleotide polymorphisms associated with an enhanced response to iloperidone during acute treatment of schizophrenia. It is considered an “atypical” antipsychotic because it displays serotonin receptor antagonism, similar to other atypical antipsychotics. The older typical antipsychotics are primarily dopamine antagonists.(3)
Iloperidone won FDA approval for use treating schizophrenia in the United States on May 6, 2009
Iloperidone (1-[4-[3-[4-(6-fluoro-1,2-benzisoxazole-3-yl)-1-piperidinyl]propoxy]-3-methoxyphenyl]ethanone) is an atypical new-generation antipsychotic medicament belonging to the class of piperidinyl-benzisoxazole derivatives, which is used to treat schizophrenia, bipolar disorder and other psychiatric conditions. Iloperidone acts as a serotonin/dopamine receptor antagonist (5-HT2A/D2).
Iloperidone, also known as Fanapt, Fanapta, and previously known as Zomaril, is an atypical antipsychotic drug used for the treatment of schizophrenia. The chemical name of iloperidone is l-[4-[3-[4-(6-fluoro-l,2-benzisoxazol-3-yl)-l- piperidinyl]propoxy] -3-methoxyphenyl]ethanone.
EP 0402644 patent discloses first synthetic route of synthesis of iloperidone as shown in Scheme I, which consists of alkylation reaction between l-(4-(3-chloropropoxy-3- methoxyphenyl)ethanone of the formula (II) and 6-fluoro-3-piperidin-4-yl-l ,2 benzisoxazole hydrochloride of the formula (III) in presence of potassium carbonate in N,N dimethyl formamide. The reaction has been subsequently worked up and the compound of formula (I) is extracted from water using ethyl acetate. The compound of formula (I) is purified by crystallization using ethanol. The overall yield of compound of formula (I) is 58%.

Formula (I)
SCHEME 1 Further, we have analyzed the reported synthetic route for synthesis of iloperidone; following limitations have been observed and identified in the reported synthetic route:
a) The yield obtained using said synthetic route as reported in US RE39198 is 58%. Hence, this route of synthesis is not cost efficient at commercial scale due to low yield;
b) Use of potassium carbonate as a base in reaction leads to formation of carbon dioxide as one of the side products during the reaction, which further hinders in the manufacturing process by actively participating in manufacturing process and thereby leads to the formation o

Formula (IV)
which is in the range of 15-20%, and thereby resulting in low yield of iloperidone;
c) Purification by crystallization using ethanol as a solvent is not effective in eliminating or controlling carbamate impurity below 0.15% as per the ICH guide lines for the known impurities; and
d) Iloperidone obtained by the above synthetic process is beige in colour.
CN101768154 discloses the synthesis of iloperidone by N-alkylation reaction between l-(4-(3- chloropropoxy-3-methoxyphenyl)ethanone of the formula (II) and 6-fluoro-3-piperidin-4-yl-l,2 benzisoxazole hydrochloride of the formula (III) in inorganic alkaline solution, particularly; alkali metal carbonate solution. We have analyzed the reported synthetic route for synthesis of iloperidone and have observed and identified that the use of alkaline carbonate solution leads to the formation of carbamate impurity in the range of 1 to 1.5%.
Several patents were published after, describing essentially the same synthetic way such as US5364866 and US5663449.
The synthesis of iloperidone is described in USRE39198 (corresponding to EP 0 402 644 example 3) according to the following synthesis scheme:

In agreement with said patent, the intermediate isolated, 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone, is reacted with 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole hydrochloride in N,N-dimethyl formamide at 90° C. for 16 hours. When the reaction is complete, the mixture is poured into water and extracted with ethyl acetate. The crude product thus obtained is crystallised twice from ethanol to give crystallised iloperidone with a total yield of 58%.
The yield of this process is very low; moreover, the process begins with two isolated intermediates, and requires an aqueous extractive work-up step with an increase in volumes and consequent reduction in the productivity and efficiency of the process. Said process also requires a double crystallisation step to obtain a beige product. The quality levels obtained are not described in the text of the example, but a beige color does not suggest a high-quality product, as iloperidone is a white substance.
The synthesis of intermediate 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone is disclosed in U.S. Pat. No. 4,366,162. Example 1 describes the preparation of said intermediate by reacting acetovanillone with 1-bromo-3-chloropropane in acetone with potassium carbonate. At the end of the reaction the resulting product is purified by distillation and obtained as an oily intermediate which is left to stand in order to obtain the solid intermediate.
The synthesis of intermediate 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone is also disclosed in U.S. Pat. No. 4,810,713. Preparation 12 describes the synthesis of said intermediate from acetovanillone and 1-bromo-3-chloropropane in sodium hydroxide alkalinized water. At the end of the reaction the product obtained is extracted in toluene, the organic phases are washed with basic aqueous solutions and finally, the intermediate 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone is crystallised with the aid of diisopropyl ether. The intermediate isolated is then recrystallised twice from cyclohexane and twice from petroleum ether.
An alternative process for the synthesis of iloperidone is reported in CN 102070626.
Scheme 2 shows the synthesis procedure:

The decision to alkylate acetovanillone with 1-chloro-3-propanol requires an extra synthesis step (to convert the OH group to an OR leaving group) compared with the procedure reported by the combination of patents USRE39198 (EP402644) and U.S. Pat. No. 4,366,162/U.S. Pat. No. 4,810,713, making said process less efficient from the economic standpoint.
WO2011061750 discloses an alternative iloperidone synthesis process as reported in Scheme 3:

Said process uses reagents such as methyl magnesium chloride to effect the Grignard reaction to convert the aldehyde group to a secondary alcohol group, which are much more complicated to manage on an industrial scale than the synthesis methods previously described. Moreover, the oxidation reaction of the next step uses reagents such as chromic acid or potassium permanganate, which have a very high environmental impact and very low industrial applicability.
WO2011055188 discloses a process for the synthesis of iloperidone comparable to the one reported in USRE39198 from two isolated intermediates 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone and 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole hydrochloride. The same patent application also gives preparation examples of the intermediate 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone isolated as crystalline solid by procedures similar to those known in the literature.
CN 101824030 reports an iloperidone synthesis method similar to that of CN 102070626 which involves the same problems of inefficiency due to the additional step of inserting the leaving group required for alkylation with 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole hydrochloride.
CN 101781243 discloses an alternative iloperidone synthesis process as reported in Scheme 4.

Said process is not advantageous compared with the preceding processes as the intermediate with the oxime group, due to the nature of this functional group, is particularly liable to degradation due to the action of numerous factors such as the presence of metals, acid pHs and basic pHs.
CN101768154 discloses a process for the synthesis of iloperidone comparable to the one reported in USRE39198 from two isolated intermediates, 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone and 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole hydrochloride.
CN 101735208 describes a process for the synthesis of iloperidone comparable to the one reported in CN 101781243, namely through the intermediate with the functional oxime group.
IN 2007MU01980 discloses a process for the synthesis of iloperidone comparable to the one reported in USRE39198 from two isolated intermediates, 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone and 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole hydrochloride.
WO 2010031497 describes an alternative iloperidone synthesis process as reported in Scheme 5.

The considerable economic disadvantage of the process reported in WO2010031497 is based on the fact that by reversing the order of alkylation and performing that of intermediate 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole hydrochloride first, a greater loss of yield is generated on this intermediate which, according to the literature, is more difficult to synthesise and consequently more expensive than the intermediate 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone, with a globally greater economic inefficiency of the iloperidone preparation process.
CN 102212063 discloses a process for the synthesis of iloperidone with the same arrangement of the synthesis steps as patent application WO 2010031497.
WO2011154860 describes a process for the synthesis of iloperidone wherein a phase transfer catalyst is used to prepare the intermediate 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone which, as in all the other preparation examples previously described, is crystallised, isolated and dried before use in the next step with 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole hydrochloride. Scheme 6 shows the synthesis scheme of the process of WO2011154860.

………………………………

……………………………………
EXAMPLE 1:
Tetrabutyl ammonium bromide (2.40 gm) was added to a stirred solution of Potassium hydroxide (0.724 kg) in mixture of Heptane (2.0L). and water (10.0L), followed by addition of 1- [4-(3-chloropropoxy)-3-methoxyphenyl]ethanone (2, 1.0kg) and 6-fluoro-3-piperidin-4-yl-l,2- benzisoxazole hydrochloride^, 1.1 1kg) at 30°C. This reaction mass was stirred for 15 to 20 min. The temperature of the reaction mass was raised to 70°C and was maintained for 8 to 10 hours. After completion of reaction (by TLC, Mobile Phase: Toluene/ Acetone/Ethyl acetate = 6:2:2 mL), the mixture was cooled to 30°C, diluted with dichloromethane (2.5 L) and stirred for 30 minutes. The dichloromethane layer was separated. The aqueous layer was re-extracted with dichloromethane (1.0L). The combined dichloromethane layer was washed with water (1.5L) and decolorized with activated charcoal (0.05 kg). The solvent was distilled off completely to obtain the residue. The residue obtained was dissolved in isopropyl alcohol (5.0L) at reflux temperature to obtain the clear solution. The clear solution obtained was cooled to 30°C followed by 0°C and stirred for 60 min to precipitate out crystals. The colorless crystals of compound (I) obtained were filtered. The crystalline solid was dried under vacuum (650-700 mm/Hg) to obtain pure compound (I) as a crystalline solid. HPLC analysis was performed for the crystalline solid obtained. The purity of Iloperidone, impurity profile and yield are shown in table 1 below.
Table 1 : Analysis data of iloperidone i.e. purity, yield and impurity profile.

EXAMPLE 2:
Tetrabutyl ammonium bromide (2.40 gm) was added to a stirred solution of Potassium hydroxide (0.724 kg) in mixture of Heptane (2.0L) and water (10.0L), followed by addition of 1- [4-(3-chloropropoxy)-3-methoxyphenyl]ethanone (2, 1.0kg) and 6-fluoro-3-piperidin-4-yl-l,2- benzisoxazole hydrochloride^, 1.1 1kg) at 30°C. This reaction mass was stirred for 15 to 20 min. The temperature of the reaction mass was raised to 70°C and maintained for 8 to 10 hours. After completion of reaction (by TLC, Mobile Phase: Toluene/ Acetone/Ethyl acetate = 6:2:2 mL), the mixture was cooled to 30°C, the reaction mixture was filtered to obtain wet crude iloperidone. Further, the obtained wet crude was dried at 60-65 °C under vacuum to furnish crude iloperidone (1.72 kg). The dried crude iloperidone was dissolved in isopropyl alcohol (5.0 L) at reflux temperature and decolorized with activated charcoal (0.05 kg). Obtained filtrate was cooled to 30°C followed by 0°C and stirred for 60 min to precipitate out crystals. The colorless crystals of compound (I) obtained were filtered. The crystalline solid was dried under vacuum (650-700 mm/Hg) to obtain pure compound (I) as a crystalline solid. HPLC analysis was performed for the crystalline solid obtained. The purity of Iloperidone, impurity profile and yield are shown in table 2 below.
Table 2: Analysis data of iloperidone i.e. purity, yield and impurity profile.

EXAMPLE-3:
……………………..
UPLC-MS [M+H+]=427
1H-NMR (in DMSO) (chemical shifts expressed in ppm with respect to the TMS signal): 2.06-1.78 (6H, m); 2.13 (2H, m); 2.49 (2H, t); 2.52 (2H, m); 2.97 (2H, m); 3.11 (1H, tt); 3.83 (3H, s); 4.12 (2H, t); 7.06 (1H, d); 7.22 (1H, m); 7.46 (1H, d); 7.61-7.58 (2H, m); 7.94 (1H, dd).
………………………………
.Improved and Efficient Process for the Production of Highly Pure Iloperidone: A Psychotropic Agent
http://pubs.acs.org/doi/full/10.1021/op400335p?prevSearch=iloperidone&searchHistoryKey=

The present work describes an improved and highly efficient process for the synthesis ofiloperidone (1), an antipsychotic agent, which is free from potential impurities. The synthesis comprises N-alkylation of 1-(4-(3-chloropropoxy)-3-methoxyphenyl)ethanone (4) with 6-fluoro-3-piperidin-4-yl-1,2-benzisoxazole hydrochloride (5) in a mixture of water and heptane as solvent and sodium hydroxide as a base in the presence of tetrabutylammonium bromide as a phase transfer catalyst to yield iloperidone (1) with a yield of around 95% and a purity of 99.80% by HPLC. The present work also describes the optimization details performed to achieve the process attributes responsible for high yield and purity.
FT-IR (KBr, λmax, cm–1): 3031, 2949, 2779, 2746, 2822, 1669, 1614, 1585, 1510, 1462, 1448, 1415, 1380, 1313, 1262, 1221, 1177, 1150, 1123, 1077, 1034, 997, 985, 955, 884, 876, 853, 812, 781, 643, 610, 569, 475.
1H NMR (CDCl3): δ 2.03–2.10 (m, 6H), 2.12–2.18 (m, 2H), 2.55–2.56 (s, 3H), 2.58–2.60 (t, 2H), 3.02–3.09 (m, 3H), 3.91 (s, 3H), 4.10–4.19 (t, 2H), 6.91–6.93 (d, 1H), 7.01–7.06 (dd, 1H), 7.21–7.24 (dd, 1H), 7.51–7.52 (d, 1H), 7.53–7.56 (dd, 1H), 7.69–7.65 (dd, 1H).
13C NMR (CDCl3): 26.02, 26.40, 30.36, 34.34, 53.36, 54.90, 55.80, 67.16, 97.04, 97.31, 110.20, 111.02, 111.98, 112.23, 117.12, 122.36, 122.46, 123.06, 130.11, 149.00, 152.66, 160.91, 162.60, 163.53, 163.66, 165.09, 198.59.
MS (ESI, m/z): 427.2 [M + H].+
Anal. Calcd (%) for C24H27FN2O4(426.48): C, 67.54; H, 6.33; found (%): C, 67.24; H, 6.18.
HPLC
HPLC analysis developed at Megafine India using a Hypersil BDS C18 column (250 mm × 4.6 mm, particle size 5 μm); mobile phase A comprising a mixture of 5.0 mM ammonium dihydrogen orthophosphate buffer and 0.1% triethylamine; mobile phase B comprising a mixture of acetonitrile/methanol in the ratio 80:20 v/v; gradient elution: time (min)/A (v/v): B (v/v); T0.01/65:35, T8.0/65:35, T25.0/35:65, T35.0/35:65, T37.0/65:35, T45.0/65:35; flow rate 1.0 mL/min; column temperature 30 °C; wavelength 225 nm. The observed retention time of iloperidone under these chromatographic conditions is about 17.0 min.
…….
http://www.asianjournalofchemistry.co.in/User/ViewFreeArticle.aspx?ArticleID=25_10_2
N oxide impurity
m.p. 155-157 ºC;
FT-IR (KBr, νmax, cm-1):
3083, 2958, 2878, 1655, 1606, 1584, 1509, 1467, 1419, 1348,1273, 1223, 1182, 1143, 1121, 1032, 971, 957, 881, 857, 813,
802;
1H NMR (300 MHz, CDCl3)
δ 1.89-1.93 (m, 2H), 2.31-2.40 (m, 2H), 2.55 (s, 3H), 2.60-2.72 (m, 2H), 3.29-3.52 (m,
2H), 3.29-3.52 (m, 2H), 3.29-3.52 (m, 2H), 3.29-3.52 (m, 1H),3.85 (s, 3H), 4.23(t, 2H, J = 6.0 Hz), 7.11 (d, 1H, J = 8.4 Hz),7.30-7.36 (m, 1H), 7.62-7.65 (m, 1H), 7.71-7.74 (dd, J = 9.3and 2.0 Hz, 1H), 8.02-8.07 (dd, J = 8.7 and 5.4 Hz, 1H);
13CNMR (75 MHz, CDCl3)
δ 22.13, 24.70, 26.35, 31.49, 55.54,63.21, 67.07, 67.82, 97.51, 110.35, 111.86, 112.67, 123.11,
123.67, 129.95, 148.63, 152.22, 160.79, 163.10, 163.69,196.40;
MS (ESI, m/z): 443 [M + H]+.
Anal. calcd. (%) forC24H27N2O5F (442.19): C, 65.15; H, 6.15; N, 6.33; found (%):C, 65.11; H, 6.09; N, 6.29.
………………………
INTERMEDIATES

FANAPT is a psychotropic agent belonging to the chemical class of piperidinyl-benzisoxazole derivatives. Its chemical name is 4′-[3-[4-(6-Fluoro-1,2-benzisoxazol-3-yl)piperidino]propoxy]-3′-methoxyacetophenone. Its molecular formula is C24H27FN2O4 and its molecular weight is 426.48. The structural formula is:
 |
Iloperidone is a white to off-white finely crystalline powder. It is practically insoluble in water, very slightly soluble in 0.1 N HCl and freely soluble in chloroform, ethanol, methanol, and acetonitrile.
|
Title: Iloperidone
CAS Registry Number: 133454-47-4
CAS Name: 1-[4-[3-[4-(6-Fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl]propoxy]-3-methoxyphenyl]ethanone
Manufacturers’ Codes: HP-873; ILO-522
Trademarks: Zomaril (Novartis)
Molecular Formula: C24H27FN2O4
Molecular Weight: 426.48
Percent Composition: C 67.59%, H 6.38%, F 4.45%, N 6.57%, O 15.01%
Literature References: Combined dopamine (D2) and serotonin (5HT2) receptor antagonist. Prepn: J. T. Strupczewski et al., EP402644; eidem, US 5364866 (1990, 1994 both to Hoechst-Roussel); eidem, J. Med. Chem. 38, 1119 (1995).
Pharmacology: M. R. Szewczak et al., J. Pharmacol. Exp. Ther. 274, 1404 (1995).
Clinical pharmacokinetics: S. M. Sainati et al., J. Clin. Pharmacol.35, 713 (1995).
HPLC determn in plasma: A. E. Mutlib, J. T. Strupczewski, J. Chromatogr. B 669, 237 (1995). Receptor binding study: S. Kongsamut et al., Eur. J. Pharmacol. 317, 417 (1996).
Review of pharmacology and therapeutic potential in schizophrenia: J. M. K. Hesselink, Curr. Opin. Cent. Peripher. Nerv. Syst. Invest. Drugs 2, 71-78 (2000); K. K. Jain, Expert Opin. Invest. Drugs 9, 2935-2943 (2000).
Properties: Crystals from ethanol, mp 118-120°.
Melting point: mp 118-120°
Therap-Cat: Antipsychotic.
Keywords: Antipsychotic; Benzisoxazoles; Serotonin-Dopamine Antagonist.
|
..
-
Bjork, A. K. K.; Abramo, A. L.; Kjellberg, B. E. S. US 4366162, 1982.
-
Strupczewski, J. T.; Helsley, G. C.; Chiang, Y.; Bordeau, K. J. EP 0402644A1, 1990.
-
Ansari, S. A.; Hirpara, H. M.; Yadav, A. K.; Gianchandani, J. P. WO2012164516, 2012.
-
Azad, M. A. K.; Pandey, G.; Singh, K.; Prasad, M.; Arora, S. K. WO2012/090138 A1, 2012.
-
Dwivedi, S. D.; Patel, D. J.; Shah, A. P. WO2012/063269, 2012.
-
Athalye, S. S.; Parghi, K. D.; Ranbhan, K. J.; Sarjekar, P. B. WO2012/153341, 2012.
-
Raman, J. V.; Rane, D.; Kevat, J.; Patil, D. WO2011/154860, 2011.
-
Reguri, B. R.; Arunagiri, M.; Yarroju, P. C.; Kasiyappan, G. S.; Ponnapall, K. WO2011/055188, 2011.
-
Shiwei, Z.; Feng, J. US 2012/0172699A1, 2012.
-
Bettoni, P.; Roletto, J.; Paissoni, P. EP 2644608A1, 2013.
-
Mathad, V. T.; Solanki, P. V.; Pandit, B. S.; Uppelli, S. B. WO2012/123963 A2, 2012.
-
Strupczewski, J. T.; Allen, R. C.; Gardner, B. A.; Schmid, B. L.; Stache, U.; Glamkowski, E. J.; Jones, M. C.; Ellis, D. B.; Huger, F. P.; Dunn, R. W. J. Med. Chem. 1985, 28, 761–769
[ACS Full Text
 ], [PubMed], [CAS]
], [PubMed], [CAS]-
20 Mucke, H. A. M.; Castañer, J. Drugs Future 2000, 25(1), 29.
-
(21) Steiner, G.; Bach, A.; Bialojan, S.; Greger, G.; Hege, H.-G.;.Höger, T.; Jochims, K.; Munschauer, R.; Neumann, B.; Teschendorf, H.-J.; Traut, M.; Unger, L.; Gross, G. Drugs Future 1998 23(2), 191.
WO2003037337A1 * Oct 29, 2002 May 8, 2003 Markus Ahlheim Depot formulations of iloperidone and a star polymer WO2008027993A2 * Aug 29, 2007 Mar 6, 2008 Eurand Inc Drug delivery systems comprising solid solutions of weakly basic drugs CN101768154A Sep 19, 2009 Jul 7, 2010 浙江华海药业股份有限公司 New preparation method of iloperidone EP0402644A1 May 16, 1990 Dec 19, 1990 Hoechst-Roussel Pharmaceuticals Incorporated N-(aryloxyalkyl)heteroarylpiperidines and -heteroarylpiperazines,a process for their preparation and their use as medicaments EP0542136A1 * Nov 5, 1992 May 19, 1993 Hoechst-Roussel Pharmaceuticals Incorporated Heteroarylpiperidines, pyrrolidines and piperazines and their use as antipsychotics and analgetics US5364866 Oct 30, 1992 Nov 15, 1994 Hoechst-Roussel Pharmaceuticals, Inc. Heteroarylpiperidines, pyrrolidines and piperazines and their use as antipsychotics and analetics US5663449 Jun 6, 1995 Sep 2, 1997 Hoechst Marion Roussel, Inc. Intermediate compounds in the synthesis of heteroarylpiperidines, pyrrolidines and piperazines USRE39198 Nov 15, 2000 Jul 18, 2006 Aventis Pharmaceuticals Inc. Heteroarylpiperidines, pyrrolidines and piperazines and their use as antipsychotics and analgesics -

Sorry, the comment form is closed at this time.