Cicaprost
94079-80-8 , as in entry 4 , J. Org. Chem. 1988,53,1227-1231
ZK-96480
phase 2
Bayer Schering Pharma (Originator)
2-
13,14-Didehydro-16,20-dimethyl-3-oxa-18,18,19,19-tetradehydro-6-carbaprostaglandin I2;
5-(7-Hydroxy-6-(3-hydroxy-4-methylnona-1,6-diynyl)-bicyclo(3.3.0)octan-3-yliden)-3-oxapentanoic acid;
2-[(2E,3aβ,6aβ)-4β-[(3S,4S)-3-Hydroxy-4-methyl-1,6-nonadiynyl]-5α-hydroxyoctahydropentalene-2-ylidene]ethoxyacetic acid;
[2-[(2E,3aβ,4S,6aβ)-4β-[(3S,4S)-3-Hydroxy-4-methyl-1,6-nonadiynyl]-5α-hydroxyoctahydropentalene-2-ylidene]ethoxy]acetic acid;
[2-[[(2E,3aS,3aβ,6aβ)-5α-Hydroxy-4β-[(3S,4S)-3-hydroxy-4-methyl-1,6-nonanediynyl]octahydropentalen]-2-ylidene]ethoxy]acetic acid;
Acetic acid, ((2E)-2-((3as,4S,5R,6as)-hexahydro-5-hydroxy-4-((3S,4S)-3-hydroxy-4-methyl-1,6-nonadiynyl)-2(1H)-pentalenylidene)ethoxy)-;
2-[2-[(2E,3aS,4S,5R,6aS)-Hexahydro-5-hydroxy-4-[(3S,4S)-3-hydroxy-4-Methyl-1,6-nonadiyn-1-yl]-2(1H)-pentalenylidene]ethoxy]acetic Acid;
Acetic acid, (2-(hexahydro-5-hydroxy-4-(3-hydroxy-4-methyl-1,6-nonadiynyl)-2(1H)-pentalenylidene)ethoxy)-, (3as-(2E,3aalpha,4alpha(3R*,4R*),5beta,6aalpha))-
| Molecular Formula | C22H30O5 |
|---|---|
| Formula Weight | 374.5 |
Prostaglandin I2 (PGI2, prostacyclin) is the most potent endogenous vasodilator that affects both the systemic and pulmonary circulation.Cicaprost is a PGI2 analog that is orally active with prolonged availabilityin vivo, having a terminal half life in plasma of one hour. In addition to their effects on smooth muscle, PGI2 analogs, including cicaprost, have been shown to inhibit the pro-
 cicaprost
cicaprost
references
1. Drugs Fut 1986, 11(11): 913
2. Synthesis of a new chemically and metabolically stable prostacyclin analogue with high and long-lasting oral activity
J Med Chem 1986, 29(3): 313
3. Journal of Organic Chemistry, 1988 , vol. 53, 6 p. 1227 – 1231 entry4
http://pubs.acs.org/doi/pdf/10.1021/jo00241a020
4. Journal of the American Chemical Society, 2003 , vol. 125, 32 p. 9653 – 9667, nmr
5. WO 2009068190
6. US 5013758
7. WO 2005009446
8. WO 1992014438
9. US2007/196510 A1
10. US2007/293552 A1
11. US2009/221549 A1
12. US2009/54473 A1
| EP0041661A2 * | May 29, 1981 | Dec 16, 1981 | Schering Aktiengesellschaft | Preparation of intermediates of carbaprostacyclines |
| EP0057660A2 * | Feb 1, 1982 | Aug 11, 1982 | Schering Aktiengesellschaft | Prostacycline derivatives, their preparation and applications as medicines |
| EP0086404A1 * | Feb 3, 1983 | Aug 24, 1983 | Schering Aktiengesellschaft | Carbacyclines, process for their preparation and their use as medicines |
| EP0086612A1 * | Feb 7, 1983 | Aug 24, 1983 | The Upjohn Company | 9-Substituted carbacyclin analogues |
| EP0087237A1 * | Feb 7, 1983 | Aug 31, 1983 | The Upjohn Company | Carbacyclin analogues |
| EP0098793A1 * | Jul 1, 1983 | Jan 18, 1984 | Schering Aktiengesellschaft | Carbacycline amides, process for their preparation and their use as medicines |
| EP0155901A1 * | Mar 6, 1985 | Sep 25, 1985 | Schering Aktiengesellschaft | Carbacyclines, process for their preparation and their use as medicines |
| EP0195379A2 * | Mar 14, 1986 | Sep 24, 1986 | G.D. Searle & Co. | Allenic prostacyclins |
| EP0195668A2 * | Mar 19, 1986 | Sep 24, 1986 | Sankyo Company Limited | Carbacyclin derivatives |
| EP0721783A1 * | Jun 6, 1995 | Jul 17, 1996 | Toray Industries, Inc. | Preventive and remedy for diseases caused by fibrinoid or thrombus formation in the lung and model animal for said diseases |
| EP2065054A1 | Nov 29, 2007 | Jun 3, 2009 | Bayer Schering Pharma Aktiengesellschaft | Combinations comprising a prostaglandin and uses thereof |
| DE3427797A1 * | Jul 25, 1984 | Feb 6, 1986 | Schering Ag | Zytoprotektive wirkung von prostacyclin-derivaten an leber, bauchspeicheldruese und niere |
| DE3448256C2 * | Jul 25, 1984 | Aug 18, 1988 | Schering Ag, 1000 Berlin Und 4709 Bergkamen, De | Cytoprotective action of prostacyclin derivatives on the pancreas |
| DE3448257C2 * | Jul 25, 1984 | Aug 18, 1988 | Schering Ag, 1000 Berlin Und 4709 Bergkamen, De | Cytoprotective action of prostacyclin derivatives on the kidney |
| DE4135193C1 * | Oct 22, 1991 | Mar 11, 1993 | Schering Ag Berlin Und Bergkamen, 1000 Berlin, De | Title not available |
| US5405870 * | Nov 4, 1993 | Apr 11, 1995 | Sankyo Company, Limited | Carbacyclin compounds; pharmaceutical compositions and method of use |
| US5489613 * | Jan 21, 1992 | Feb 6, 1996 | Sankyo Company, Limited | Carbacyclin derivatives, process for their preparation and compositions containing them |
| US5716989 * | Nov 27, 1991 | Feb 10, 1998 | Schering Aktiengesellschaft | Bicyclo 3.3.0!octane derivatives, process for their production and their pharmaceutical use |
| US5891910 * | Jun 6, 1995 | Apr 6, 1999 | Schering Aktiengesellschaft | 9-halogen-(Z) prostaglandin derivatives, process for their production and their use as pharmaceutical agents |
| US6040336 * | Aug 6, 1996 | Mar 21, 2000 | Schering Aktiengesellschaft | Prostane derivatives and the combination thereof with antibiotics in the treatment of bacterial infections |
| US6225347 | Sep 27, 1994 | May 1, 2001 | Schering Aktiengesellschaft | 9-halogen-(Z)-prostaglandin derivatives, process for their production and their use as pharmaceutical agents |
| WO1986000808A1 * | Jul 18, 1985 | Feb 13, 1986 | Schering Ag | Prostacycline derivatives with a cytoprotective action on the liver, the pancreas and the kidney |
| WO1987005294A1 * | Mar 9, 1987 | Sep 11, 1987 | Schering Ag | Cyclodextrinclathrates of carbacycline derivatives and their use as medicinal drugs |
| WO1988001867A1 * | Sep 1, 1987 | Mar 24, 1988 | Schering Ag | Topical agent containing prostacycline derivatives |
| WO1991014675A1 * | Mar 27, 1991 | Sep 29, 1991 | Schering Ag | Bicyclo[3.3.)]octane derivatives, process for producing them and their pharmaceutical use |
| WO1992014438A2 * | Feb 11, 1992 | Aug 13, 1992 | Schering Ag | Prostacycline and carbacycline derivatives as agents for treating feverish complaints |
| WO1994003175A1 * | Aug 9, 1993 | Feb 17, 1994 | Schering Ag | Use of prostane derivatives of formulae i and ii for the production of a medicament for the treatment of chronic polyarthritis |
| WO1997006806A1 * | Aug 6, 1996 | Feb 27, 1997 | Schering Ag | Use of prostane derivatives and the combination thereof with antibiotics in the treatment of bacterial infections |
 cicaprost
cicaprost
…………………………
Journal of Organic Chemistry, 1988 , vol. 53, 6 p. 1227 – 1231 entry4
http://pubs.acs.org/doi/pdf/10.1021/jo00241a020
ZK 96 480 (4). A solution of 19 (68 mg, 0.13 mmol) in eth-
er-toluene (3 mL, 2:l) was added to tetrabutylammonium hydrogen sulfate containing HzO (2 drops). After adding 50% aqueous NaOH (0.8 mL), the whole reaction mixture was stirred at 55 “C for 48 h. The reaction was quenched with HzO, acidified with 5% aqueous HC1, extracted with ethyl acetate, washed withH20 and brine, and concentrated to give ZK 96 480 (4) (42 mg, 86%) as a colorless viscous oil:
[alZzD +138.25O (c 1.025, CHCI,). see pdf file for correct cut paste
Other spectral data were identical with those of an authentic
sample.’
(1) Skuballa, W.; Schillinger, E.; Stiinebecher, C.-St.; Vorbriiggen, H.
J. Med. Chem. 1986,29, 313.
……………………………….
Skuballa, W.; Schillinger, E.; Stiinebecher, C.-St.; Vorbriiggen, H.
J. Med. Chem. 1986,29, 313.
http://pubs.acs.org/doi/pdf/10.1021/jm00153a001
see original pdf file for structures
we replaced the methylene group in the
3-position of 1, iloprost by an oxygen atom to prevent the 6-oxi-
dation of the upper side chain. The resulting decrease in
intrinsic activity was compensated for by modification of
the lower side chain. We converted the 13,14-double bond
into a triple bond, introduced a further methyl group at
(2-20, and synthesized selectively the pure 16(S)-methyl
diastereomer. These modifications resulted in the struc-
ture of 2 cicaprost (ZK 96 480), a carbacyclin analogue with a bio-
logical activity at least as high as that of prostacyclin and
iloprost.
The synthesis of 2 started with the preparation of the
lower side chain by resolving racemic 2-methyl-4-heptynoic
acid (3).7 By application of the method of Helmchen et
al.,” 3 was converted with phosphorus trichloride into the
acid chloride 4, which gave with D-(-)-a-phenylglycinol a
pair of diastereomeric amides. After chromatographic
separation on SOz, the more polar amide 5 (mp 124 ‘C)
was hydrolyzed with 3 N H2S04 in dioxane to furnish the
optically pure 2s-configurated acid 6 ([a]D -1.2’ (c 1,
EtOH), bp 128 ‘C (12 mm)). The 2s configuration of 6
was determined by hydrogenation of 6 to 2(S)-methyl-
heptanoic acid ([“ID +17.7′ (c 1, EtOH)), which was com-
pared with 2-methyl-alkanoic acids of known absolute
config~ration.~ Esterification of 6 with diazomethane
followed by reaction of the methyl ester 7 ([a]D +12.2’ (c
1, EtOH), bp 70 ‘C (12 mm)) with the lithium salt of ethyl
methylphosphonate afforded the optically pure phospho-
nate 8 ([‘Y]D +35.3’ (c 1, EtOH), bp 123 “C (0.3 mm)).
Condensation of the phosphonate 8 with the readily
available optically pure bicyclic aldehyde 93,4 (NaH, DME,
-20 “C) in the presence of N-bromosuccinimide furnished
the a,P-unsaturated bromo ketone 10 in 60% yield: oil;
(3 H, d, J = 7 Hz, CHCH,), 3.91 (4 H, m, OCH2CH20), 5.21
(1 H, m, H-llp), 7.09 (1 H, d, J = 10 Hz, H-13), 7.42-7.92
(5 H, m, COPh); IR (neat) 1720 (COPh), 1690 (COC=C)
cm-‘. Reduction of 10 (NaBH,, CH,OH, -40 “C) gave a
ca. 1:l mixture of the allylic alcohols lla and llb, which
was separated chromatographically.’O Dehydrobromina-
tion (50% aqueous NaOH, toluene, catalytic NBu4/HS04,
25 “C) of the less polar alcohol lla with concomitant sa-
ponification of the benzoate group followed by acidic
(HOAc, H20) cleavage of the ketal moiety afforded the
ketone 12 (73% from lla): oil; ‘H NMR (CD2C12) 6 1.06
(3 H, d, J = 6.8 Hz, CHCH,), 1.10 (3 H, t, J = 7.5 Hz,
CH,CH,), 4.22 (1 H, m, H-llb), 4.38 (1 H, m, H-158); IR
(neat) 1730 (C=O) cm-‘. After silylation of the hydroxyl
groups in 12 (C1SiMe2-t-Bu, DMF, imidazole), the ketone
13 was subjected to a Horner-Wittig reaction with triethyl
phosphonoacetate (KO-t-Bu, THF, 0 “C). Reduction of
the 1:l mixture of the isomeric a,p-unsaturated esters 14
with diisobutylaluminum hydride (toluene, 0 “C) gave after
chromatographic separation the E isomer 15a (32% from
12) and the less polar 2 isomer 15b.11
Etherification of 15a under phase-transfer conditions
with tert-butyl bromoacetate (50% aqueous NaOH, tolu-
ene, catalytic Bu4NHS04, 25 “C) was accompanied by
simultaneous cleavage of the tert-butyl ester to give 16
(87%). Finally, removal of the silyl ether groups (tetra-
n-butylammonium fluoride, THF, 25 “C) afforded 2 cicaprost, in
86% yield: oil;
‘H NMR (CD,Cl,) 6= delta 1.07 (3 H, d, J = 6.8
Hz), 16@-CH3), 1.11 (3 H, t, J = 7.5 Hz, CH2CH3), 3.97 (1
H, m, H-llP), 4.06 (2 H, m, OCH,CO), 4.12 (2 H, m, =
H, m, H-5); IR (neat) 1730 (COOH) cm-‘.
………………

An asymmetric synthesis of the anti-metastatic prostacyclin analogue cicaprost and a formal one of its isomer isocicaprost by a new route are described. A key step of these syntheses is the coupling of a chiral bicyclic C6−C14 ethynyl building block with a chiral C15−C21 ω-side chain amide building block with formation of the C14−C15 bond of the target molecules.
A highly stereoselective reduction of the thereby obtained C6−C21 intermediate carrying a carbonyl group at C15 of the side chain was accomplished by the chiral oxazaborolidine method. The chiral phosphono acetate method was used for the highly stereoselective attachment of the α-side chain to the bicyclic C6−C21 intermediate carrying a carbonyl group at C6.
Asymmetric syntheses of the bicyclic C6−C14 ethynyl building blocks were carried out starting from achiral bicyclic C6−C12 ketones by using the chiral lithium amide method. In the course of these syntheses, a new method for the introduction of an ethynyl group at the α-position of the carbonyl group of a ketone with formation of the corresponding homopropargylic alcohol was devised.
Its key steps are an aldol reaction of the corresponding silyl enol ether with chloral and the elimination of a trichlorocarbinol derivative with formation of the ethynyl group. In addition, a new aldehyde to terminal alkyne transformation has been realized. Its key steps are the conversion of an aldehyde to the corresponding 1-alkenyl dimethylaminosulfoxonium salt and the elimination of the latter with a strong base.
Two basically different routes have been followed for the synthesis of the enantiomerically pure C15−C21 ω-side chain amide building block. The first is based on the chiral oxazolidinone method and features a highly stereoselective alkylation of (4R)-N-acetyl-4-benzyloxazolidin-2-one, and the second encompasses a malonate synthesis of the racemic amide and its efficient preparative scale resolution by HPLC on a chiral stationary phase containing column
…….
https://www.google.co.in/patents/EP0119949A1
(5E) -13,14,18,13,19,19-Hecadehydro-3-oxa-6a-carba-prostaglandin I 2derivatives of the general formula I

(5E) – (16S) -13,14-didehydro-16 ,20-dimezhyl-3-oxa-18 ,18,19,19-tetradehydro-6a-carbaprostaglandin 1 2
- Example 1(5E) – (16S) -13,14-didehydro-16 ,20-dimezhyl-3-oxa-18 ,18,19,19-tetradehydro-6a-carbaprostaglandin 1
2
-
[0028]To a solution of 0.4 g in 12 ml of tetrahydrofuran was added to 80 mg of 55% sodium hydride (in mineral oil) and cook for 1 hour reflux. Is added to a solution of 127 mg of bromoacetic in 4 ml of tetrahydrofuran, boiled under reflux for 18 hours, diluted with ether and extracted four times with 30 ml of 5% sodium hydroxide. This extract is adjusted with 10% sulfuric acid at 0 ° C to pH 3 and extracted with methylene chloride. The organic extract is shaken with brine, dried over magnesium sulfate and evaporated under vacuum. Obtained 220 mg hydropyranyläther), which are for the elimination of the protective groups is stirred for 18 hours with 15 ml of acetic acid / water / tetrahydrofuran (65/35/10) at 25 ° C. It is evaporated to the addition of toluene, and the residue is chromatographed on silica gel with ethyl acetate / 0.1 – 1% acetic acid. This gives 145 mg of the title compound as a colorless Ö1.
-
[0029]IR (CHC1 3): 3600, 3400 (broad), 2 93 0 222 3, 1730, 1600, 1425, 1380/cm.
-
[0030]The starting material for the above title compound is prepared as follows:
1 a)
-
[0031]To a suspension of 3.57 g of sodium hydride (55% in mineral oil) in 360 ml of dimethoxyethane was added dropwise at O ° C, a solution of 21.9 g of 3-methyl-2-oxo-oct-5-in-phosphonsäuredimethyl esters in 140 ml of dimethoxyethane was stirred for 1 hour at 0 ° C and then add 14.56 g of finely powdered N-bromosuccinimide. It is stirred for 1 hour at O ° C, treated with a solution of 22.5 g of (lR, 5S, 6R, 7R) -3,3 – ethylenedioxy-7-benzoyloxy-6-formyl-bicyclo [3.3.0] octane in 180 ml of dimethoxyethane and 4 hours the mixture is stirred at 0 ° C. The reaction mixture is diluted with 3 1 ether, washed neutral with brine, dried with sodium sulfate and evaporated in vacuo. The residue is chromatographed with hexane / ether as eluent on silica gel. Following three chromatography of the respective diastereomeric mixed fractions obtained as polar compound 8.1 g and a polar compound 7.4 g of the title compound as colorless oils.
-
[0032]IR: 2935, 2878, 17 15, 1690, 1601, 1595, 1450, 1270, 948/cm.
1 b)
-
[0033]To a solution of 7.4. G of produced according to Example 1 a) ketone in 140 ml of methanol is added at -20 ° C. 3 g of sodium borohydride in portions and stirred for 30 minutes at -20 ° C. Then diluted with ether, washed neutral with water, dried over magnesium sulfate and evaporated under vacuum.
-
[0034]The crude product (15-epimer) is dissolved in 300 ml of methanol, added to 2.95 g of potassium carbonate and stirred for 21 hours at 23 ° C under argon. Then concentrated in vacuo, diluted with ether and washed neutral with brine. It is dried over magnesium sulfate and evaporated under vacuum. By column chromatography on silica gel with ether / methylene chloride (7 +3) first obtained 2.6 g of the 15SS-configured alcohol as well as 2.1 g of the more polar component 15a-configured alcohol (PG nomenclature) as colorless oils.
-
[0035]A solution of 2.1 g of the above prepared alcohol 15a, 20 mg of p-toluenesulfonic acid and 1.4 g of dihydropyran in 50 ml of methylene chloride is stirred for 30 minutes at 0 ° C. Then it is poured into dilute sodium bicarbonate solution, extracted with ether, washed neutral with water, dried over magnesium sulfate and evaporated under vacuum. Chromatography of the residue on silica gel, using hexane / ether (6 +4), 2.6 g of the title compound as a colorless oil.
-
[0036]IR: 2939, 2877, 1450, 969, 948 / cm.
1 c) bicyclo [3.3.0] octane-3-one
-
[0037]A solution of 290 mg of the of Example 1 b) the compound prepared in 2.5 ml of dimethyl sulfoxide and 1 ml of tetrahydrofuran is mixed with 112 mg of potassium tert-butoxide and stirred for 2 hours at 23 ° C. It is diluted with 10 ml of water and extracted three times with 10 ml of ether / hexane (7 +3), wash the extract with water until neutral, dried over brine and evaporated under vacuum.
-
[0038]It is stirred for 22 hours with the residue 15 ml of acetic acid / water / tetrahydrofuran (65/35/10) evaporated in a vacuum with the addition of toluene, and the residue is purified by chromatography on silica gel. With ether eluted 150 mg oily substance, which is reacted in 5 ml of dichloromethane with 140 mg of dihydropyran and 1 mg of p-toluenesulfonic acid at 0 ° C.. After 30 minutes, diluted with ether, extracted with 5% sodium bicarbonate solution and brine, dried over magnesium sulfate and evaporated under vacuum. Chromatography of the residue on silica gel with hexane / ether (1 +1), 185 mg of the title compound as a colorless oil.
-
[0039]IR: 2940, 2876, 2216, 1738, 1020, 970 / cm.
1 d)
-
[0040]To a solution of 529 mg Phosphonoessigsäuretri acid ethyl ester in 10 ml of tetrahydrofuran is added at 0 C 225 mg of potassium tert-butoxide, stirred for 10 minutes, treated with a solution of 0.6 g of the product of Example 1 c) ketone in 6 ml of toluene and stirred for 22 hours at 23 ° C. It is diluted with 150 mL of ether, shake once with water, once with 20% sodium hydroxide, washed neutral with water, dried over magnesium sulfate and evaporated under vacuum. The residue is filtered using hexane / ether (6 +4) over silica gel. Thereby obtain 0.58 g of the unsaturated ester as a colorless oil.
-
[0041]IR: 2940, 2870, 2212, 1704, 1655, 970 / cm.
-
[0042]It adds 150 mg of lithium aluminum hydride in portions at 0 ° C to a stirred solution of 570 mg of the ester prepared in 25 ml of ether and stirred for 30 minutes at 0 ° C. Destroying the excess reagent by dropwise addition of ethyl acetate, added to 1 ml of water, stirred for 3 hours at 20 ° C, filtered and evaporated under vacuum. The residue is chromatographed with ether / hexane (3 +2) on silica gel. Thereby obtained as a non-polar compound 140 mg of 2 – {(Z) – (1S, 5S, 6S, 7R) -7 – (tetrahydropyran-2-yloxy) -6 – / R3S, 4S)-4-methyl-3-( tetrahydropyran-2-yloxy)-nona-1 ,6-diinyl]-bicyclo [3.3.0] octane-3-ylidene} – ethane-1-ol and 180 mg of the title compound as a colorless oil.
-
[0043]IR: 3620, 3450 (broad), 2940, 2860, 2212, 970/cm.
…………….

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family

 iloprost
iloprost In 2003, CoTherix licensed exclusive rights from Schering AG to market iloprost in the U.S. for primary pulmonary hypertension while Schering AG retained rights to the product outside the U.S. In April 2005, CoTherix established a collaborative research and development agreement with Quadrant to develop an extended-release formulation of iloprost inhalation solution. Iloprost was designated as an orphan medicinal product for the treatment of pulmonary hypertension in December 2000 by the EMEA and will fall under orphan drug protection until 2013.
In 2003, CoTherix licensed exclusive rights from Schering AG to market iloprost in the U.S. for primary pulmonary hypertension while Schering AG retained rights to the product outside the U.S. In April 2005, CoTherix established a collaborative research and development agreement with Quadrant to develop an extended-release formulation of iloprost inhalation solution. Iloprost was designated as an orphan medicinal product for the treatment of pulmonary hypertension in December 2000 by the EMEA and will fall under orphan drug protection until 2013. ILOPROST
ILOPROST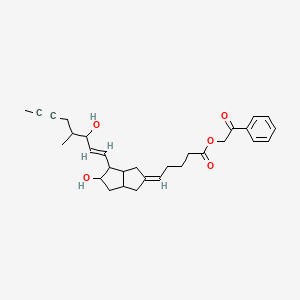 iloprost phenacyl ester
iloprost phenacyl ester

















































 11.25 M in NH3)], Na2CO3 [15% w/w solution (4 L)]. More EtOAc (4 L) was added, and the organic layer was washed with water (4 L). The organic phase was then concentrated to 2.5 L; again fresh EtOAc (4 L) was added, and the solution was concentrated to 2.5 L to give a solution of casopitant 2.
11.25 M in NH3)], Na2CO3 [15% w/w solution (4 L)]. More EtOAc (4 L) was added, and the organic layer was washed with water (4 L). The organic phase was then concentrated to 2.5 L; again fresh EtOAc (4 L) was added, and the solution was concentrated to 2.5 L to give a solution of casopitant 2.