












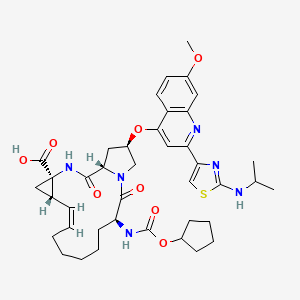
CILUPREVIR
(1S,4R,6S,7Z,14S,18R)-14- {[(cyclopentyloxy)carbonyl]amino}-18-[(7-methoxy-2- {2-[(propan-2-yl)amino]-1,3-thiazol-4-yl}quinolin-4- yl)oxy]-2,15-dioxo-3,16- diazatricyclo[14.3.0.0{4,6}]nonadec-7-ene-4- carboxylic acid
Ciluprevir, BILN-2061, BILN 2061, CHEBI:161337, BILN2061, BILN 2061ZW, BILN-2061-ZW,
CAS , 300832-84-2

Ciluprevir is used in the treatment of hepatitis C. It is manufactured by Boehringer Ingelheim Pharma GmbH & Co. KG under the research code of BILN-2061. It is targeted against NS2-3 protease.[1]
Ciluprevir is an HCV NS3 protease inhibitor which had been in phase II clinical trials at Boehringer Ingelheim for the treatment of hepatitis C, however, no recent developments from the company have been reported.
1. Challenge and Opportunity in Scaling-Up Metathesis Reaction: Synthesis of Ciluprevir (BILN 2061)Peter J. Dunn, et al
http://onlinelibrary.wiley.com/doi/10.1002/9781118354520.ch10/summary
DOI: 10.1002/9781118354520.ch10
2. Synthesis of BILN 2061, an HCV NS3 protease inhibitor with proven antiviral effect in humans
Org Lett 2004, 6(17): 2901
http://pubs.acs.org/doi/full/10.1021/ol0489907
3. Efficient synthesis of (S)-2-(cyclopentyloxycarbonyl)-amino-8-nonenoic acid: Key buiding block for BILN 2061, an HCV NS3 protease inhibitor
Org Process Res Dev 2007, 11(1): 60
4. Chinese Journal of Chemistry, 2011 , vol. 29, 7 pg. 1489 – 1502
DOI: 10.1002/cjoc.201180270
…………………………..
US 8222369
WO 2006071619
WO 2000059929
WO 2004092203
WO 2004039833
WO 2004037855
WO 2006036614
WO 2006033878
WO 2005042570
WO 2004093915
………………………………………………………………………..
https://www.google.co.in/patents/US8222369


…………………………………………………………………..
http://www.google.com/patents/WO2000059929A1
COMPD 822 IS CILUPREVIR IN TABLE 8
EXAMPLE 8 Synthesis of 4-hydroxy-7-methoxy-2[4(2-isopropylaminothiazolyl)] quinoline (8f ) Note: [ A variety of 2-alkylaminothiazolyl substituents were made using the same synthetic scheme where compound 8b was replaced by other alkyl thioureas.]


8b 8c 8d

A. The protocol used for the conversion of -anisidine to 8a was identical to that described in the literature: F.J. Brown et al. J. Med. Chem. 1989, 32 , 807-826. However, the purification procedure was modified to avoid purification by chromatography. The EtOAc phase containing the desired product was treated with a mixture of MgSO4, charcoal and 5% w/w (based on expected mass) silica gel. After filtration on celite, the product was triturated with ether. Compound 8a was obtained as a pale brown solid in >99% purity (as confirmed by HPLC).
B. A suspension of isopropyl thiourea (8b, 3.55 g, 30 mmol) and 3- bromopyruvic acid (8c, 5 g, 1 eq.) in dioxane (300 mL , 0.1 M) was heated to 80 °C.
Upon reaching 80 C the solution became clear and soon after the product precipitated as a white solid. After 2 hours of heating, the solution was cooled to RT and the white precipitate was filtered to obtain compound 8d in high purity (>98% purity as confirmed by NMR) and 94% yield (7.51 g). C. A mixture of the carboxylic acid 8d (4.85 g, 18.2 mmol) and the aniline derivative 8a (3 g, leq.) in pyridine (150 mL, 0.12 M) was cooled to -30 °C (upon cooling, the clear solution became partially a suspension). Phosphorus oxychloride (3.56 ml, 2.1 eq.) was then added slowly over a 5 min period. The reaction was stirred at -30 C for 1 h, the bath was removed and the reaction mixture was allowed to warm-up to RT. After 1.5 h the reaction mixture was poured into ice, the pH was adjusted to 11 with aqueous 3N NaOH, extracted with CH2C12, dried over anhydrous MgSO4, filtered and concentrated under vacuum. The beige solid was then purified by flash chromatography (45% EtOAc in hexane) to give compound 8e as a pale yellow solid in 73% yield (6.07 g). D. A solution of tBuOK (2.42 g, 21.6 mmol) in anhydrous tBuOH (40ml, 0.14 M, distilled from Mg metal) was heated to reflux. Compound 8e (1.8g, 5.4 mmol) was added portion-wise over 5 min and the dark red solution formed was stirred at reflux for an additional 20 min (completion of the reaction was monitored by HPLC). The mixture was cooled to RT and HCl was added (4 N in dioxane, 1.5 eq.). The mixture was then concentrated under vacuum, in order to assure that all of the
HCl and dioxane were removed, the product was re-dissolved twice in CH2C12 and dried under vacuum to finally obtain the HCl salt of compound 8f as a beige solid (1.62 g, 93% pure by HPLC). The product was then poured into a phosphate buffer
(IN NaH2PO4, pH=~4.5) and sonicated. The beige solid was filtered and dried under vacuum to give compound 8f (1.38 g, 81% yield) as a beige solid (91% pure by HPLC).
*H NMR (400 MHz, DMSO) δ 8.27 (s, IH), 8.12 (d, IH, J = 9.2 Hz), 7.97 (br.s, IH), 7.94 (s, IH), 7.43 (s, IH), 7.24 (dd, IH, J = 9.2, 2.2 Hz), 3.97 (m, IH), 3.94 (s, 3H), 1.24 (d, 2H, J = 6.4 Hz)
…………
METHYL ESTER
EXAMPLE 34c
Using the same procedure as described in example 34 but reacting bromoketone 34f with commercially available N-iso-propylthiourea gave # 822

Η NMR (400 MHz, DMSO-d6) δ 8.63 (s, IH), 8.33-8.23 (bs, IH), 8.21 (d, J = 9.2 Hz, IH), 8.04 (d, J = 8.3 Hz, IH), 7.86 (bs, IH), 7.77 (s, IH), 7.35-7.23 (m, 2H), 5.81 (bs, IH), 5.52 (dd, J = 8.5 Hz, IH), 5.27 (dd, J = 9.2 Hz, IH), 4.65 (d, J = 11.8 Hz, IH), 4.51 (dd, J = 7.6 Hz, IH), 4.37 (bs, IH), 4.15 (bs, IH), 4.07-3.98 (m, 2H), 3.97 (s, 3H), 3.88 (d, J = 8.9 Hz, IH), 2.60-2.53 (m, 2H), 2.47-2.37 (m, 2H), 2.19-2.10 (dd, J = 9.2 Hz, IH), 1.80-1.64 (m, 2H), 1.63-1.29 (m, 13H), 1.27 and 1.25 (2 x d, J – 6.5 Hz, 6H), 1.23-1.09 (m, 2H). MS; es+: 775.0 (M + H)+, es : 772.9 (M – H)\
CILUPREVIR IS FREE ACID OF ABOVE AND HAS ENTRY 822 TABLE 8
………
FREE AMINO COMPD
(Table 8)




A. To a solution of the macrocyclic intermediate 23b (13.05 g, 27.2 mmol, 1.0 eq.), Ph3P (14.28 g, 54.4 mmol, 2.0 eq) and 2-carboxymethoxy-4-hydroxy-7- methoxyquinoline (WO 00/09543 & WO 00/09558) (6.67 g, 28.6 mmol, 1.05 eq) in
THF (450 mL) at 0°C, DIAD (10.75 mL, 54.6 mmol, 2.0 eq) was added dropwise over a period of 15 min. The ice bath was then removed and the reaction mixture was stirred at RT for 3 h. After the complete conversion of starting material to products, the solvent was evaporated under vacuum, the remaining mixture diluted with
EtOAc, washed with saturated NaHCO3 (2x) and brine (lx), the organic layer was dried over anhydrous MgSO4, filtered and evaporated to dryness. Pure compound 34a was obtained after flash column chromatography; the column was eluted first with hexane/EtOAc (50:50), followed by CHCl3/EtOAc (95:5) to remove Ph3PO and
DIAD byproducts and elution of the impurities was monitored by TLC. Finally, the desired product 34a was eluted from the column with CHC13/ EtOAc (70:30).
Usually, the chromatography step had to be repeated 2-3 times before compound 34a could be isolated in high purity as a white solid with an overall yield of 68% (12.8 g, 99.5% pure by HPLC).
B. To a solution of the Boc-protected intermediate 34a (1.567g) in CH2C12 (15 mL), 4N HCl in dioxane (12 mL) was added and the reaction mixture was stirred at RT for 1 h. [In the event that a thick gel would form half way through the reaction period, an additional 10 mL CH2C12 was added.] Upon completion of the deprotection the solvents were evaporate to dryness to obtain a yellow solid and a paste like material. The mixture was redissolved in approximately 5% MeOH in
CH2C12 and re-evaporated to dryness under vacuum to obtain compound 34b as a yellow solid, which was used in the next step without any purification. C. To a solution of cyclopentanol (614 μL, 6.76 mmoL) in THF (15 mL), a solution of phosgene in toluene (1.93 M, 5.96 mL, 11.502 mmol) was added dropwise and the mixture was stirred at R.T. for 2 h to form the cyclopentyl chloroformate reagent (z). After that period, approximately half of the solvent was removed by evaporation under vacuum, the remaining light yellow solution was diluted by the addition of CH2C12 (5 mL) and concentrated to half of its original volume, in order to assure the removal of all excess phosgene. The above solution of the cyclopentyl chloroformate reagent was further diluted with THF (15 mL) and added to the amine-2HCl salt 34b. The mixture was cooled to 0 C in an ice bath, the pH was adjusted to -8.5-9 with the addition of Et3N (added dropwise) and the reaction mixture was stirred at 0 C for 1 h. After that period, the mixture was diluted with
EtOAc, washed with water (lx), saturated NaHCO3 (2x), H2O (2x) and brine (lx).
The organic layer was dried over anhydrous MgSO4, filtered and evaporated under vacuum to obtain a yellow-amber foam. Compound 34c was obtained as a white foam after purification by flash column chromatography (using a solvent gradient from 30% hexane to 20% hexane in EtOAc as the eluent) in 80% yield (1.27 g) and >93% purity. D. The dimethyl ester 34c (1.17g) was dissolved in a mixture of
THF/MeOH/H2O (20 mL, 2:1:1 ratio), and an aqueous solution of NaOH (1.8 mL,
IN, 1 eq.) was added. The reaction mixture was stirred at RT for 1 h before it was evaporated to dryness to obtain the sodium salt 34d as a white solid (-1.66 mmol). Compound 34d was used in the next step without purification.
E. The crude sodium salt 34d (1.66 mmoL) was dissolved in THF (17 mL), Et3N was added and the mixture was cooled to 0 C in an ice bath. Isobutylchloroformate
(322 μl, 2.5 mmol) was added dropwise and the mixture was stirred at 0 C for 75 min. After that period, diazomethane (15 mL) was added and stirring was continued at 0 C for 30 min and then at RT for an additional 1 h. Most of the solvent was evaporated to dryness under vacuum, the remaining mixture was diluted with EtOAc, washed with saturated NaHCO3 (2x), H2O (2x) and brine (lx), dried over anhydrous MgSO4, filtered and evaporated to dryness to obtain compound 34e as a light yellow foam (1.2g, -1.66 mmol). The diazoketone intermediate 34e was used in the next step without purification.
F. The diazoketone 34e (1.2g, 1.66 mmoL) dissolved in THF (17 mL) was cooled to 0 C in an ice bath. A solution of aqueous HBr (48%, 1.24 mL) was added dropwise and the reaction mixture was stirred at 0 C for 1 h. The mixture was then diluted with EtOAc, wash with saturated NaHCO3 (2x), H2O (2x) and brine (lx), the organic layer was dried over anhydrous MgSO4, filtered and evaporated to dryness to obtain the β-bromoketone intermediate 34f as a light yellow foam (-1.657 mmol).
G. To a solution of the bromoketone 34f (600 mg,0.779 mmol) in isopropanol (5 mL), thiourea (118 mg, 1.55 mmol) was added and the reaction mixture was placed in a pre-heated oil bath at 75 C where it was allowed to stir for 1 hr. The isopropanol was then removed under vacuum and the product dissolved in EtOAc
(100 mL). The solution was washed with saturated NaHCO3 and brine, the organic layer was dried over anhydrous Na2SO4, filtered and evaporated to afford the crude product 34g (522 mg) as a red-brown solid. This material was used in the final step without any further purification.
H. The crude methyl ester 34g (122 mg, 0.163 mmol) was dissolved in a solution of THF/MeOH/H2O (2:1:1 ratio, 4 mL) and saponified using LiOH»H2O (89 mg, 2.14 mmol). The hydrolysis reaction was carried out over a 12-15 h period at RT. The solvents were then removed under vacuum and the crude product purified by C18 reversed phase HPLC, using a solvent gradient from 10% CH3CN in H2O to 100%
CH3CN, to afford the HCV protease inhibitor #812 as a yellow solid (24 mg, 20% overall yield for the conversion of intermediate 34f to inhibitor #812).
*H NMR (400 MHz, DMSO-d6) δ 8.63 (s, IH), 8.26-8.15 (m, 2H), 7.79 (bs, IH), 7.72
(bs, IH), 7.50 (bs, 2H), 7.33-7.25 (m, 2H), 5.77 (bs, IH), 5.52 (dd, J = 8.3 Hz, IH), 5.27 (dd, J = 9.2 Hz, IH), 4.64 (d, J = 10.8 Hz, IH), 4.50 (dd, J = 8.3 Hz, IH), 4.39-4.31 (m, IH), 4.08-3.99 (m, 2H), 3.94 (s, 3H), 3.87 (d, J = 9.5 Hz, 2H), 2.65-2.53 (m, 2H), 2.46- 2.36 (m, 2H), 2.20-2.12 (dd, J = 8.6 Hz, IH), 1.80-1.64 (m, 2H), 1.63-1.06 (m, 14H). MS; es+: 733.2 (M + H)+, es“: 731.2 (M – H)\
………………..
http://www.google.com/patents/WO2006036614A2
(Z)-( 1S,4R, 14S, 18R)- 14-Cyclopentyloxycarbonylamino- 18-[2-(2- isopropylamino-thiazol-4-yl)-7-methoxy-quinolin-4-yloxy]-2,15-dioxo-3,16-diaza- tricyclo[14.3.0.0 ‘ ]nonadec-7-ene-4-carboxylic acid , whose chemical structure is as follows:

, provided for in Tsantrizos et al., U.S. Patent No. 6,608,027 Bl,
…………………………
https://www.google.co.in/patents/WO2005090383A2
ENTRY 218

…………………..
http://www.google.com/patents/WO2004039833A1

……………..
A new procedure for the practical synthesis of (S)-2-(cyclopentyloxycarbonyl)amino-8-nonenoic acid, a key building block for BILN 2061, an HCV NS3 protease inhibitor, has been developed. The key step features a kinetic resolution of racemic 2-acetylamino-8-nonenoic acid with acylase I. In addition, the undesired (R)-2-acetylamino-8-nonenoic acid was recycled after racemization. The procedure was implemented for the production of (S)-2-(cyclopentyloxycarbonyl)amino-8-nonenoic acid on pilot-plant scale.


……………
nmr
Synthesis of BILN 2061, an HCV NS3 protease inhibitor with proven antiviral effect in humans
Org Lett 2004, 6(17): 2901
http://pubs.acs.org/doi/full/10.1021/ol0489907
http://pubs.acs.org/doi/suppl/10.1021/ol0489907/suppl_file/ol0489907si20040715_032207.pdf procedure
http://pubs.acs.org/doi/suppl/10.1021/ol0489907/suppl_file/ol0489907si20040715_032254.pdf nmr spectra
BILN 2061:
Methyl ester 18 (2.69 g, 3.41 mmol) was dissolved in a mixture of THF
(40 mL), MeOH (20 mL) and water (20 mL) and added LiOH.H2O (1.14 g, 27.3 mmol).The resulting mixture was left to stir at RT for 15 h. The solvents were then removedunder reduced pressure and the crude product was redissolved with EtOAc and dilutedwith brine. The pH of the aqueous layer was adjusted to 6 with aqueous HCl (1N) and theaqueous phase was extracted with EtOAc (3x). The combined organic phase werewashed with water, brine, dried over MgSO4 and concentrated under reduced pressure toafford BILN 2061 as a yellow solid (2.63 g, 99% yield). HPLC(A) 99%, MS m/z (ES+)773 (M+H)+, (ES-) 775 (M-H)-;
1H NMR (DMSO-d6) δ 8.63 (s, 1H), 8.26-8.15 (m, 2H),
7.79 (bs, 1H), 7.72 (bs, 1H), 7.50 (bs, 2H), 7.33-7.25 (m, 2H), 5.77 (bs, 1H), 5.52 (dd, J=8.3 Hz, 1H), 5.27 (dd, J= 9.2 Hz, 1H), 4.64 (d, J= 10.8 Hz, 1H), 4.50 (dd, J= 8.3 Hz, 1H),4.39-4.31 (m, 1H), 4.08-3.99 (m, 2H), 3.94 (s, 3H), 3.87 (d, J= 9.5 Hz, 2H), 2.65-2.53(m, 2H), 2.46-2.36 (m, 2H), 2.20-2.12 (dd, J= 8.6 Hz, 1H), 1.80-1.64 (m, 2H), 1.63-1.06(m, 14H); HRMS calcd for C40H51N6O8S: 775.3489; found: 775.3476
…………………………
| WO2007019674A1 | Aug 3, 2006 | Feb 22, 2007 | Boehringer Ingelheim Int | Viral polymerase inhibitors |
| WO2010021717A2 * | Aug 20, 2009 | Feb 25, 2010 | Sequoia Pharmaceuticals, Inc. | Hcv protease inhibitors |
| WO2010080874A1 | Jan 7, 2010 | Jul 15, 2010 | Scynexis, Inc. | Cyclosporine derivative for use in the treatment of hcv and hiv infection |
| EP1455809A2 * | Dec 13, 2002 | Sep 15, 2004 | Bristol-Myers Squibb Co. | Inhibitors of hepatitis c virus |
| EP2364984A1 | Aug 28, 2006 | Sep 14, 2011 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases |
| EP2366704A1 | Aug 28, 2006 | Sep 21, 2011 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases |
| US7368452 | Jul 18, 2006 | May 6, 2008 | Enanta Pharmaceuticals, Inc. | Quinoxalinyl macrocyclic hepatitis C serine protease inhibitors |
| US7608590 | Jan 28, 2005 | Oct 27, 2009 | Medivir Ab | HCV NS-3 serine protease inhibitors |
| US7671032 | Jan 28, 2005 | Mar 2, 2010 | Medivir Ab | HCV NS-3 serine protease inhibitors |
| US7816348 | Jan 29, 2007 | Oct 19, 2010 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| US7897622 | Aug 10, 2007 | Mar 1, 2011 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| US8148399 | Jul 28, 2006 | Apr 3, 2012 | Tibotec Pharmaceuticals Ltd. | Macrocyclic inhibitors of hepatitis C virus |
| US8153800 | Aug 3, 2011 | Apr 10, 2012 | Tibotec Pharmaceuticals Ltd. | Macrocyclic inhibitors of hepatitis C virus |
| US8242140 | Jul 31, 2008 | Aug 14, 2012 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| US8349869 | Mar 6, 2012 | Jan 8, 2013 | Tibotec Pharmaceuticals Ltd. | Macrocylic inhibitors of hepatitis C virus |
| US8476257 | Dec 3, 2008 | Jul 2, 2013 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| US8541402 | May 3, 2012 | Sep 24, 2013 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| WO2000059929A1 * | Apr 3, 2000 | Oct 12, 2000 | Boehringer Ingelheim Ca Ltd | Macrocyclic peptides active against the hepatitis c virus |
| WO2003053349A2 * | Dec 13, 2002 | Jul 3, 2003 | Squibb Bristol Myers Co | Inhibitors of hepatitis c virus |
| WO2003064455A2 * | Jan 24, 2003 | Aug 7, 2003 | Boehringer Ingelheim Ca Ltd | Macrocyclic peptides active against the hepatitis c virus |
| WO2003066103A1 * | Feb 5, 2003 | Aug 14, 2003 | Boehringer Ingelheim Pharma | Pharmaceutical compositions for hepatitis c viral protease inhibitors |

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family

One Response to “CILUPREVIR”
Sorry, the comment form is closed at this time.
[…] CILUPREVIR » All About Drugs […]