











![]()
Department of Chemistry, University of Louisville
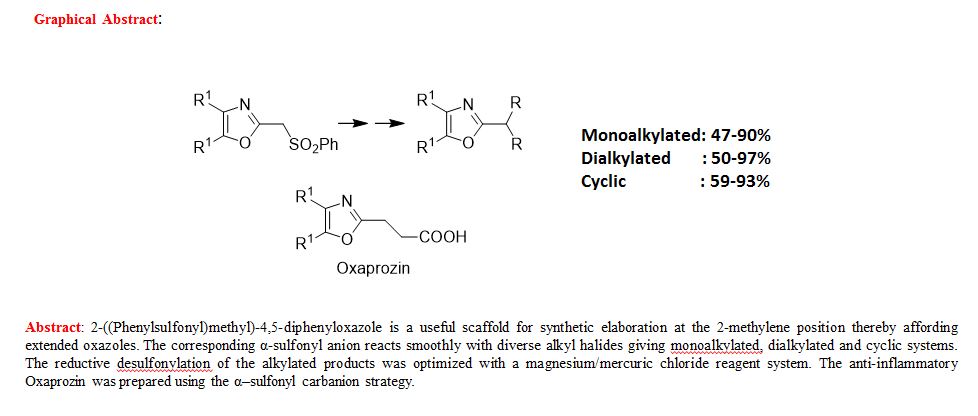
Synthesis of Extended Oxazoles III: Reactions of 2-(Phenylsulfonyl)methyl-4,5-Diaryloxazoles
Pravin C. Patil and Frederick A. Luzzio*
Department of Chemistry, University of Louisville, 2320South Brook Street, Louisville, Kentucky 40292
Faluzz01@louisville.edu
*Corresponding Author: Email: faluzz01@louisville.edu
J Org. Chem., 2016, 81(21), pp 10521–10526.
Publication Date (Web): July 21, 2016 (Note)
DOI: 10.1021/acs.joc.6b01280

Frederick A. Luzzio
Professor, Organic Chemistry: Organic and Medicinal Chemistry

Typical Procedure for Aluminum/HgCl2-Mediated Desulfonylation for Synthesis of 4 (Eq. 1) and 18 (Table 2). To a solution of the alkylated 2-(sulfonylethyl)-4,5-diphenyloxazole 5 (0.12 mmol, 1.0 equiv) and crystals of mercuric chloride (0.034 mmol, 0.3 equiv), in methanol (15 mL), was added an excess of food-grade aluminum foil (2.32 mmol, 20 equiv) with vigorous stirring under a nitrogen atmosphere. The resulting heterogeneous mixture was heated at reflux until the metal disappeared. The reaction mixture was then allowed to cool to room temperature and filtered through a Celite bed followed by washing with methanol (2 x 15mL). The filtrate was concentrated to a crude residue which was submitted to gravity-column chromatography on silica gel to provide 2-methyl-4,5-diphenyloxazole 4 (96%) or 2-ethyl-4,5-diphenyloxazole 18 (97%).
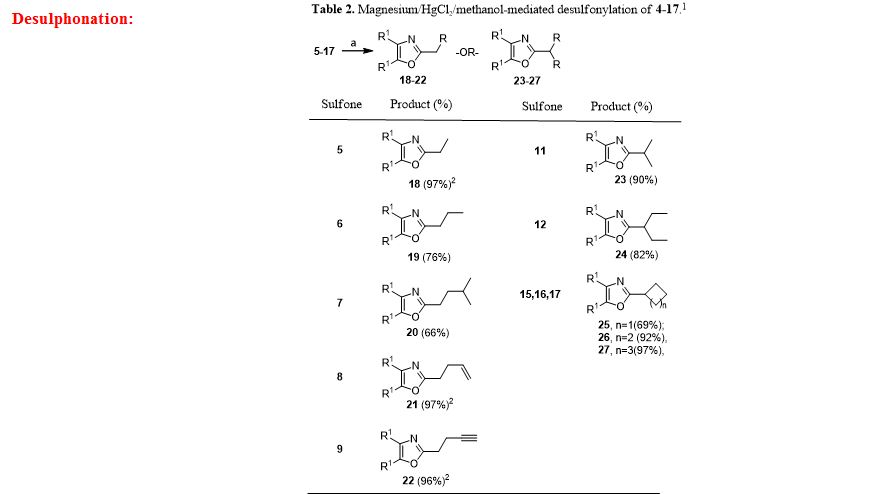
General procedure for Magnesium/HgCl2-Mediated Desulfonylation of Alkylated Sulfones 5-17. To a stirred solution of an alkylated 2-(phenylsulfonyl)methyl-4,5-diphenyloxazole (0.12 mmol, 1.0 equiv. from Table 1) in methanol (5 mL) was added magnesium turnings (1.73 mmol, 15 equiv) and crystals of mercuric chloride (0.012 mmol, 0.1 equiv) at room temperature. The reaction mixture was stirred at room temperature (2 h) while monitoring the reaction progress by TLC. After the reaction was complete, the reaction mixture was filtered through a Celite bed followed by washing with methanol (2 x 10 mL). The filtrate was concentrated and the resultant crude residue was submitted to gravity-column chromatography on silica gel (hexane/ethyl acetate) to afford the pure products 18–27 listed in Table 2.
Typical Procedure for Aluminum/HgCl2-Mediated Desulfonylation for Synthesis of 4 (Eq. 1) and 18 (Table 2). To a solution of the alkylated 2-(sulfonylethyl)-4,5-diphenyloxazole 5 (0.12 mmol, 1.0 equiv) and crystals of mercuric chloride (0.034 mmol, 0.3 equiv), in methanol (15 mL), was added an excess of food-grade aluminum foil (2.32 mmol, 20 equiv) with vigorous stirring under a nitrogen atmosphere. The resulting heterogeneous mixture was heated at reflux until the metal disappeared. The reaction mixture was then allowed to cool to room temperature and filtered through a Celite bed followed by washing with methanol (2 x 15mL). The filtrate was concentrated to a crude residue which was submitted to gravity-column chromatography on silica gel to provide 2-methyl-4,5-diphenyloxazole 4 (96%) or 2-ethyl-4,5-diphenyloxazole 18 (97%).
General procedure for Magnesium/HgCl2-Mediated Desulfonylation of Alkylated Sulfones 5-17. To a stirred solution of an alkylated 2-(phenylsulfonyl)methyl-4,5-diphenyloxazole (0.12 mmol, 1.0 equiv. from Table 1) in methanol (5 mL) was added magnesium turnings (1.73 mmol, 15 equiv) and crystals of mercuric chloride (0.012 mmol, 0.1 equiv) at room temperature. The reaction mixture was stirred at room temperature (2 h) while monitoring the reaction progress by TLC. After the reaction was complete, the reaction mixture was filtered through a Celite bed followed by washing with methanol (2 x 10 mL). The filtrate was concentrated and the resultant crude residue was submitted to gravity-column chromatography on silica gel (hexane/ethyl acetate) to afford the pure products 18–27 listed in Table 2.

Typical procedure: Synthesis of Oxaprozin
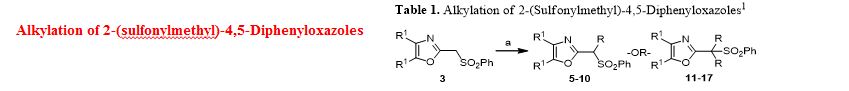
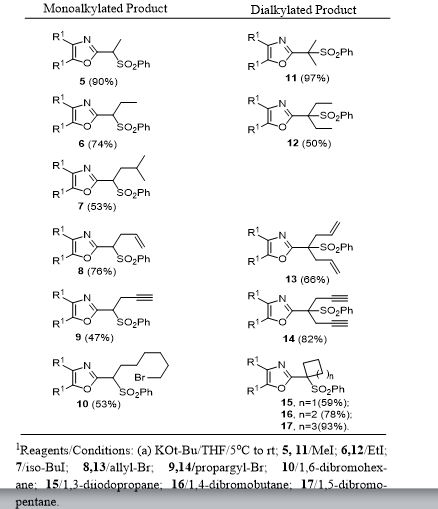
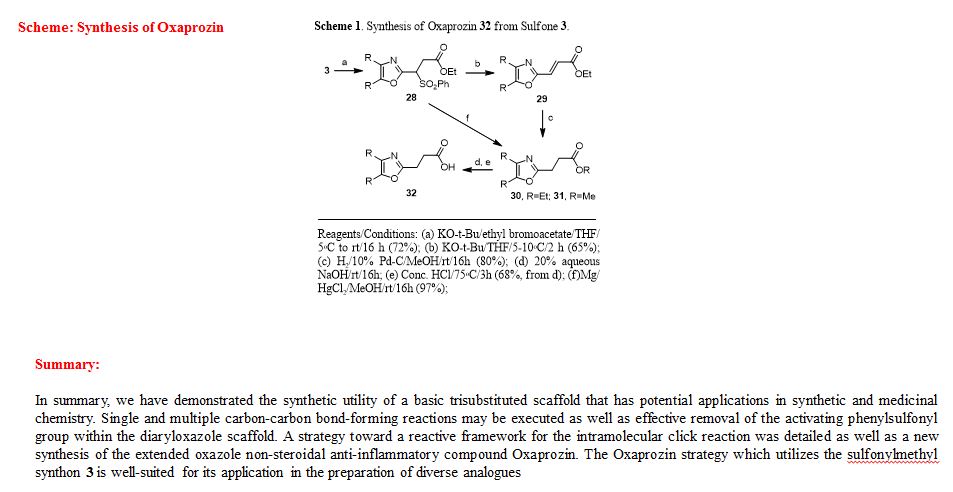
Ethyl 3-(4,5-diphenyloxazol-2-yl)-3-(phenylsulfonyl)propanoate (28). To a prechilled solution of 2-(phenylsulfonyl)methyl-4,5-diphenyloxazole 3 (100 mg, 0.27 mmol) in dry THF (15 mL) was added potassium tert-butoxide (33 mg, 0.29 mmol) under a nitrogen atmosphere. The resulting yellow solution was stirred (5°C) for 30 min. To the reaction mixture was slowly added ethyl bromoacetate (49 mg, 32.4 μL, 0.29 mmol) and stirring was continued (16 h) at room temperature. Upon completion of reaction as indicated by TLC, the reaction mixture was quenched with cold water (20 mL) and extracted with dichloromethane (2 x 20 mL). The organic layers were combined, dried over anhydrous sodium sulfate and concentrated to obtain a crude oily residue. The residue was submitted to gravity-column chromatography on silica gel (hexane/ethyl acetate, 4:1) afford pure ethyl 3-(4,5-diphenyloxazol-2-yl)-3-(phenylsulfonyl)propanoate 28 as off-white solid ( 88 mg, 72%).
Ethyl 3-(4,5-diphenyloxazol-2-yl)acrylate (29). To a cooled (5°C) solution of sulfonyloxazole ester 28 (225 mg, 0.49 mmol) in dry THF was added potassium tert-butoxide (60.2 mg, 0.54 mmol) under nitrogen and the reaction mixture was then stirred at 5-10°C (2 h) while monitoring by TLC. After completion of the reaction, the reaction mixture was extracted with dichloromethane (2 x 25 mL) followed by washing the extracts with water and brine then drying over anhydrous Na2SO4. Removal of the drying agent and concentration of the filtrate gave a crude residue which was submitted to gravity-column chromatography (hexane/ethylacetate, 4:1) to provide unsaturated oxazole ester 29 as a colorless oil (100 mg, 65%).
Ethyl 3-(4,5-diphenyloxazol-2-yl)propanoate (30).17 The unsaturated oxazole ester 30 (160 mg, 0.50 mmol) was dissolved in methanol (25 mL) then 10% Pd/C (16 mg, 10% wt/wt) was added at room temperature. The reaction mixture was purged with nitrogen while stirring followed by the addition of hydrogen gas (balloon) and then stirring was continued (16 h) under an atmosphere of hydrogen. Upon completion of reaction, the reaction mixture was filtered through a bed of Celite while washing with methanol (2 x 30 mL). The combined filtrates were concentrated and the crude residue was submitted to gravity-column chromatography (hexane/ethyl acetate, 4:1) to afford 30 as an off-white solid (129 mg, 80%).
Methyl 3-(4,5-diphenyloxazol-2-yl)propanoate (31).13 To a clear solution of sulfonyloxazole ester 28 (80 mg, 0.173 mmol) in methanol (10 mL) was added magnesium turnings (63 mg, 2.60 mmol) followed by solid mercuric chloride (4.7 mg, 0.017 mmol) at room temperature. The resulting reaction mixture was stirred (2 h) while monitoring the reaction progress by TLC. After completion of the reaction, the heterogeneous mixture was then filtered through a Celite bed followed by washing with methanol (2 x 15 mL). The methanolic filtrates were combined and concentrated to afford a crude residue. The residue was submitted to gravity-column chromatography (hexane/ethylacetate, 4:1) to provide ester 31 as an off-white solid (52 mg, 97%).
3-(4,5-Diphenyloxazol-2-yl)propanoic acid (Oxaprozin) (32).13 Ethyl ester 30 (128 mg, 0.39 mmol) or methyl ester 31 (65 mg, 0.21 mmol) and 20% aquous NaOH solution (3 mL) was stirred overnight at room temperature. Upon completion of reaction as indicated by TLC, the reaction mixture was slowly acidified to pH 3-4 using conc. HCl (3 mL) at room temperature and stirring was continued (3 h). After the neutralization was complete the reaction mixture was diluted with cold water (15 mL) and extracted with dichloromethane (2 x 15 mL). The organic extracts were combined, dried over anhydrous Na2SO4 and concentrated to give a white solid residue. The residue was submitted to gravity-column chromatography (chloroform/methanol, 9:1) to afford pure Oxaprozin 32 as white solid (80 mg, 68%, from the ethyl ester 30) or (60 mg, 97%, from the methyl ester 31).
ABOUT GUEST BLOGGER

Dr. Pravin C. Patil
Postdoctoral Research Associate at University of Louisville
see…….http://oneorganichemistoneday.blogspot.in/2017/04/dr-pravin-patil.html
Dr. Pravin C Patil completed his B.Sc. (Chemistry) at ASC College Chopda (Jalgaon, Maharashtra, India) in 2001 and M.Sc. (Organic Chemistry) at SSVPS’S Science College Dhule in North Maharashtra University (Jalgaon, Maharashtra, India) in year 2003. After M.Sc. degree he was accepted for summer internship training program at Bhabha Atomic Research Center (BARC, Mumbai) in the laboratory of Prof. Subrata Chattopadhyay in Bio-organic Division. In 2003, Dr. Pravin joined to API Pharmaceutical bulk drug company, RPG Life Science (Navi Mumbai, Maharashtra, India) and worked there for two years. In 2005, he enrolled into Ph.D. (Chemistry) program at Institute of Chemical Technology (ICT), Matunga, Mumbai, aharashtra, under the supervision of Prof. K. G. Akamanchi in the department of Pharmaceutical Sciences and Technology.
After finishing Ph.D. in 2010, he joined to Pune (Maharashtra, India) based pharmaceutical industry, Lupin Research Park (LRP) in the department of process development. After spending two years at Lupin as a Research Scientist, he got an opportunity in June 2012 to pursue Postdoctoral studies at Hope College, Holland, MI, USA under the supervision of Prof. Moses Lee. During year 2012-13 he worked on total synthesis of achiral anticancer molecules Duocarmycin and its analogs. In 2014, he joined to Prof. Frederick Luzzio at the Department for Chemistry, University of Louisville, Louisville, KY, USA to pursue postdoctoral studies on NIH sponsored project “ Structure based design and synthesis of Peptidomimetics targeting P. gingivalis.
During his research experience, he has authored 23 international publications in peer reviewed journals and inventor for 4 patents.
Prof K. G. Akamanchi

SEE…………
Frederick A. Luzzio
Professor, Organic Chemistry: Organic and Medicinal Chemistry
- 502-852-7323
- 502-852-6066
- faluzz01@louisville.edu
About
The long term goals of our research are focused at the interface of chemistry and biology. We are interested in solving problems in biomedicine using the techniques and application of synthetic organic, medicinal and natural products chemistry. Toward our goals in biomedicine we concentrate our efforts in the following three areas of organic chemistry: (1) the development of new methods and strategy which are applicable to the synthesis of biologically active compounds; (2) the total synthesis of a wide range of complex molecules including natural products, pharmaceutical leads and their analogues; and (3) the isolation and discovery of biologically active compounds from natural sources. Within our objectives in item 1 (above), we have had a long-term collaboration with the Clinical Pharmacology Section of the National Cancer Institute in which we have synthesized metabolites and analogues of thalidomide, a small-molecule immunomodulator and angiogenesis inhibitor. The derivatives and analogues of thalidomide were stereospecifically synthesized in order to ascertain the mode of action and the molecular target of this small molecule. Ultimately, the synthetic studies are leading to analogues of thalidomide which are more potent, but which have less undesirable side effects than the parent compound. In the neurosciences area we have completed an enantioselective synthesis of both optical isomers of a key intermediate in preparing the histrionicotoxins, a group of compounds which are isolated for the neurotoxic Amazon “poison dart” frogs. One of our present natural products projects (under item 3,above) entails the isolation, neurotoxicity assays and synthesis of a series of naturally-occurring compounds called acetogenins from the North American paw paw tree Asimina triloba. The isolation, purification and structural confirmation of the natural products has been conducted in collaboration with the Neurosciences Department within the University of Louisville School of Medicine. In the area of anti-infectives (under 1), we are designing and synthesizing an array of nitrogen and nitrogen/oxygen heterocyclic scaffolds bearing acetylenic and azido groups for use in the so-called “click reaction.” The multiply-connected scaffolds have proven to be effective for inhibiting micro-organisms working in tandem to produce biofilms necessary for their establishment and survival.
1976 B.S. Vanderbilt University
1979 M.S. Tufts University
1982 Ph.D. Tufts University
1982-1985 Postdoctoral Fellow, Harvard University
Current Service
Executive Committee/Treasurer, International Society of Heterocyclic Chemistry HETCHEM@louisville.edu
Links
Gordon Research Conferences on Natural Products 2009
The Natural Products Gordon Conference. 1951-2011
International Society of Heterocyclic Chemistry
///////